












【佳学基因检测13】医院所需要的下一代测序技术(NGS测序)
三甲医院及临床医学高通量基因检测技术导读
下一代测序 (NGS) 是许多实验室用来检测遗传性疾病和dota2吧雷电竞 突变的技术。这项技术对于许多执业病理学家来说是新的,他们可能不熟悉 NGS 的用途、方法和局限性。
科谱写作目的
让病理学家熟悉 NGS 的几个方面,包括当前和扩展的用途;方法学,包括实验室操作步骤工作方面、生物信息学和解释;验证和熟练程度;限制;以及与将 NGS 数据整合到患者护理中相关的问题。
科普内容收集
该评论基于同行评审的文献和在主要学术中心的临床环境中使用 NGS 的个人经验。
临床所需的高通量、下一代测序技术应用共识
随着技术、生物信息学和资源的发展,NGS 的临床应用将会增加,以解决局限性并提高结果质量。临床实验室面临的挑战是确保测试具有临床相关性、成本效益,并且可以整合到临床护理中。
新一代测序 (NGS) 或大规模平行测序是一种同时对数百万个 DNA 片段(或互补 DNA)进行测序的方法,由于它能够同时分析多个基因或基因区域,因此在临床实验室中得到了迅速采用。与传统方法相比,单次测试。与任何新技术一样,NGS 在临床实验室中的使用已经发展并将随着时间的推移继续发展。该技术的新应用正在继续开发,新的生物信息学和湿工作台技术正在开发中,以解决当前的限制并提高性能,并且正在积累关于罕见变异解释的新知识。本文概述了临床 NGS,包括近期趋势以及在不久的将来可能发生的演变。该评论基于同行评审的文献和在主要学术中心的临床环境中使用 NGS 的个人经验。明尼苏达大学费尔维尤医学中心的分子诊断实验室自 2012 年以来提供了一种基于捕获的 NGS 遗传病检测,涵盖了 568 个基因,并在 2014 年扩展到了 2484 个基因。此外,自 2014 年以来,我们提供了用于肿瘤学(血液系统恶性肿瘤和实体瘤)的 21 基因热点 NGS 面板。 我们的实验室每年检测约 800 例 NGS 遗传病和 800 例 NGS 肿瘤病例,两位作者签署了其中约三分之二的病例。第一作者还参加了一个国家病理学组织的委员会,其中讨论和解决了 NGS 相关问题。
NGS的当前和扩展用途
在许多临床实验室中,二代测序是种系(遗传)和体细胞(获得性突变)基因突变的既定测试方法。对于遗传性疾病,种系突变检测可能包括靶向 panel、全外显子组、全基因组或线粒体 DNA 测序。 针对各种遗传性疾病(例如免疫缺陷、骨髓衰竭综合征、失明、耳聋、线粒体疾病、肾脏疾病、神经系统疾病、结缔组织疾病、心肌病、和dota2吧雷电竞 易感综合征等。 与临床表型相关的基因的靶向 panel 通常是遗传性疾病检测的第一线,而全外显子组测序则保留用于靶向检测无法提供信息的病例。 全外显子组测试通常涉及测试孩子和父母双方(三人组测试)以帮助解释变异。此外,NGS 技术还用于分析产前环境中的游离 DNA。
用于癌症检测的靶向试剂盒也因实验室而异。 靶向组可能很广泛,包括实体和血液系统恶性肿瘤的基因,或者可能更专注于特定类型的恶性肿瘤(如髓系肿瘤)。 panel 中的任何给定基因都可以是有效测序的或仅部分测序的(例如热点区域)。对于种系和体细胞测试,在决定使用测试时了解目标面板的内容非常重要。目前临床上并未将全外显子组和全基因组测序用于肿瘤学检测。
NGS 的一些新应用贼近已进入临床领域或正在积极研究用于临床用途,包括循环肿瘤 DNA 检测、人类白细胞抗原 (HLA) 分型、微生物分析、RNA 测序和表达以及甲基化。NGS 的这些新用途中的一些可能得益于现在可用的新仪器的独特优势(参见“新仪器”部分)。使用 NGS 进行 HLA 分型有一些挑战需要克服:区分低频等位基因和高频伪影,以及将 2 个相似等位基因区分为 2 个不同的等位基因。 然而,较新的数据分析技术(例如逐步阈值聚类)已允许将 NGS 作为 HLA 分型的临床选择进行探索。 使用 NGS 进行短串联重复序列 (STR) 的同一性测试会遇到与其他重复区域相同的问题(参见下文难以测序的区域);然而,更新的数据分析技术再次在解决这个问题上取得了进展,并且可能适用于其他重复区域。临床 NGS 的其他用途包括药物遗传学、微生物测序和高级血型分型(例如 A1 型与 A2 型)。对这些主题的进一步讨论超出了本文的范围。
一段时间以来,无细胞 DNA 已被用于产前检测。然而,循环肿瘤 DNA (ctDNA) 的 NGS,即肿瘤衍生的无细胞 DNA,是一项较新的发展,现已在临床上可用。 这种检测通常被称为液体活检。测序 ctDNA 的潜在应用包括癌症筛查或诊断、监测进展或反复,以及指导已知癌症诊断患者的治疗。大多数研究都评估了 ctDNA 测序检测已知癌症患者体细胞突变的能力以及监测疾病的能力。多项研究表明,通过对 ctDNA 测序来监测已知突变与疾病的反复/进展相关。 此外,使用 ctDNA 突变检测来帮助指导已知肿瘤患者的治疗已显示出实用性,例如,酪氨酸激酶抑制剂对肺癌表皮生长因子受体 ( EGFR ) 激活突变的反应。尽管 ctDNA 检测突变的敏感性可能低于检测肿瘤组织,但 ctDNA 贼常见的临床应用似乎是用于转移性癌症患者,因为没有足够的组织进行检测,并且重复活检会导致显着的发病率和死亡率,并且当检测 ctDNA 是一个合理的选择时。 使用 ctDNA 筛查或诊断早期癌症存在更多问题。大多数对已知癌症患者的研究并未包含正常对照,但有限数量的靶向测序研究显示正常对照中存在一定程度的突变检测(假阳性),尽管通常处于低水平。 检测早期癌症(假阴性)的敏感性低是另一个限制。研究表明,早期肿瘤的敏感性在 30% 至 60% 范围内,并且某些肿瘤类型的假阴性率可能更高,因为 ctDNA 似乎由于细胞凋亡和坏死而释放。 这些假阳性和假阴性问题限制了 ctDNA 在早期癌症诊断或筛查中的实际应用。
目前临床NGS的方法
实验室操作步骤
样品经过 DNA 提取、文库制备、靶标富集和测序(图 1,A 和 B)。

图1:A,基于捕获的测序的实验室操作步骤概述。DNA 进行文库制备,然后在测序前进行基于捕获的选择。B,基于聚合酶链反应 (PCR) 的测序的湿式工作台步骤概述。PCR 选择步骤发生在文库制备之前,或者可以与基于 PCR 的测序中的文库制备步骤结合使用。
DNA 提取
几乎所有的 DNA 提取方法都是可以接受的。福尔马林固定、石蜡包埋 (FFPE) 组织的提取方法可能需要特别小心,在某些情况下可能需要宏观解剖或显微解剖以富集肿瘤。 DNA 定量由 Qubit 或 Picogreen(Thermo Fisher Scientific,Waltham,Massachusetts)而不是标准分光光度法进行。
文库制备
文库制备是指制备用于测序仪的 DNA 的过程。尽管有许多方法可用,但它们都导致将 DNA 分解成片段并在末端添加接头。 适配器可能包括分子条形码(以允许合并患者样本)、通用聚合酶链式反应 (PCR) 引物、将 DNA 片段与表面结合的杂交序列以及启动测序的识别位点。术语库是指这些带有侧翼接头的 DNA 片段,可用于测序。接头之间的 DNA 片段大小称为插入片段大小. 刀片尺寸可能不同,短刀片尺寸和长刀片尺寸有不同的优势。较短的片段更有可能两端落在外显子内,这通常是感兴趣的区域,而较长的片段更有可能在内含子中有 1 个末端,如果仅外显子区域被检测,这可能会增加结构重排的检测。选择(图2)。有关结构重排的更多详细信息,请参阅结构变异和拷贝数变异。

图2:具有短 DNA 插入片段(顶部)的片段更有可能有两个配对末端读数(红色条)落在外显子内。具有长插入大小的片段更有可能跨越重排的断点,这通常发生在内含子中。转载自 Yohe SL。热点话题聚焦——临床二代测序的新前沿。
目标测序区域富集
生成的文库经过富集以进行全外显子组分析和靶向测试,或直接测序以进行全基因组分析。可以通过与互补序列杂交(序列捕获)或通过 PCR 进行富集。PCR 富集通常与文库制备步骤相结合,因为选择感兴趣区域的引物也可能包含接头序列。富集策略的选择通常由临床应用决定:序列捕获更适合大基因组区域,PCR 更适合需要更大富集的较小区域。
测序
大多数临床测序是在两种主要仪器中的一种上进行的:Illumina 测序仪(加利福尼亚州圣地亚哥),包括 HiSeq、MiSeq 和 NexSeq;或 Ion Torrent 系列机器,包括 IonPGM、IonProton 和 IonS5(Thermo Fisher Scientific)。这两种类型的机器在化学成分、检测方法、优缺点方面有所不同 (表 1)。
表格1。
Illumina 和 Ion Torrent 平台的比较
| 测序平台 | 局部克隆扩增 | 侦查 | 读取长度、基数 | 优点 | 缺点 |
| Illuminaa | 流动池 | 发荧光的 | 100–300 | 成对末端读取 | 富含GC区域中的错误 |
| Iron Torrent | 珠粒和乳液 | 离子(pH) | 100–400 | 短期运行时间 | 均聚物误差 |
| 成对两端测序 | 截断误差 |
Illumina 和 Ion Torrent 平台的第一个测序步骤是固定每个 DNA 片段并对其进行克隆扩增。需要克隆扩增来产生足够大的检测信号。Ion Torrent 使用微珠乳液进行固定和克隆扩增,而 Illumina 测序仪使用流通池。 流动槽或珠子包含与 DNA 片段上的部分接头杂交的序列。输入 DNA 浓度对于确保每个珠子仅结合 1 个 DNA 片段并确保 DNA 片段在流动槽上的间距良好至关重要。克隆扩增步骤产生一个珠子或簇,其中包含大约 1000 个与其他分子物理分离的独特亲本 DNA 分子的相同拷贝。对于 Ion Torrent,然后将珠子放入孔中(每孔 1 个珠子)。
Illumina 测序仪使用带荧光检测的合成测序( 图3 , A 到 D)。所有 4 个荧光标记的核苷酸都被添加并竞争下一个空间。互补标记的核苷酸将结合,但阻断剂阻止每轮添加超过 1 个核苷酸(可逆终止化学)。剩余的未结合核苷酸被洗掉。激光激发导致记录的荧光发射(同时为每个 DNA 片段簇)。荧光标签和阻断剂被切割,然后下一轮开始。在每一轮中,从每个 DNA 簇中读取 1 个碱基对。这个过程可以在 DNA 片段的另一端重复,称为配对末端读数(表 2)。
和Ion Torrent基于离子的测序(E)说明.jpg)
图 3:Illumina 边合成边测序(A 到 D)和 Ion Torrent 离子测序(E)的图示。A,荧光标记的核苷酸(黑色圆圈和彩色圆圈)竞争 DNA 链上的下一个互补空间(灰色圆圈)。B,当掺入荧光标记的核苷酸时,它会阻止核苷酸的进一步添加。C,清洗流通池,去除额外的荧光标记核苷酸,激光信号导致荧光发射。D,荧光标签和阻断剂被去除并洗掉,允许在下一个循环中掺入下一个碱基。这同时发生在簇中的所有 DNA 链和流动槽上的所有簇中。E,在每个循环中,以一组模式添加一个碱基。对于此示例,碱基添加的顺序是 A、T、C 和 G,然后重复。如果加入碱基,则会释放离子,从而导致与连续添加的碱基数量成正比的 pH(电压)变化。
表 2。
| 名词 | 解释 |
| 对齐 | 将读取的序列与另一个序列进行比较,并确定其所属位置。有两种类型的对齐:重新组装或重新排序。 |
| 从头组装 | 将序列读取与该样本的所有其他序列读取进行比较,以确定一致性序列 |
| 重新测序 | 将读取的序列与参考序列(例如,参考人类基因组)进行比较。也称为映射 |
| 鱼饵 | 能够靶向感兴趣序列(例如互补DNA或RNA)的人工构建体 |
| 序列),并可用于分离该目标序列。用于序列捕获目标富集。 | |
| Demultiplex解复用 | 通过唯一标识符代码将单个样本的读取与多个样本的合并读取分开在合并之前附加的接头。 |
| 地图/地图 | 将读取的序列与引用进行比较,并确定其所属位置。另见对准, |
| 重新排序。 | |
| 阅读 | 可以参考单个碱基对位置的序列结果或单个碱基的序列结果 |
| 从单个克隆扩增DNA簇读取碱基对的序列长度。 |
定义
Ion Torrent 测序不同,因为每轮仅添加一个碱基(例如,第一轮中的 A,第二轮中的 T)。当加入添加的碱基时,会释放氢离子,同时检测到孔内每个珠子的 pH 值变化;如果没有加入碱基,则不会产生电压。并入多于 1 个相同的碱基会导致成比例地更高的电压信号,高达约 6 到 8 个碱基(图3 ,E )。如果包含超过 6 到 8 个碱基,则信号不再成比例,并且无法确定确切的数量。
生物信息学
从任何一种仪器读取的原始数据都经过一系列生物信息学过程(也称为管道),贼终提供变体调用文件 (VCF(表 3 )。这些过程包括多路分解(表 2)、质量分析、将读数映射到参考基因组(重测序)和变体识别/注释。由于这些专业流程,可能需要专门的生物信息学人员来建立和维护临床 NGS 服务。
表3:下一代测序的文件类型|
文件类型 |
全名 |
描述 |
近似文件大小(平均覆盖率1603) 4800基因外显子组 |
|
|
FASTQ BAM |
具有序列和变异一致性评估的文件 序列比对/映射的二进制版本 变量调用文件 |
解复用后的原始排序数据 对齐后的数据排序 |
50 GB 16 GB |
18 GB 6 GB |
| VCF | 包含相对于引用调用的变体的文件 | 9.3 GB | 3.5 MB | |
使用条形码标记样本的 DNA 片段可以将多个样本汇集在一起进行测序,从而降低测序成本。然而,这个过程需要一个多路分解步骤,其中所有读取在进一步分析之前按条形码/样本排序。具有原始读取的多路分解文件称为FASTQ 文件(表 3)。
在解复用之后,将样品的单个读数映射(表 2)到参考基因组(BAM 文件表 3),并记录参考和测序读数之间的任何差异。对于全基因组测序或序列捕获,相同(重复)读数会被丢弃,但对于基于扩增子的测序则不会。如果多个读数显示相同的差异,则称为变体(所需读数的数量或百分比的阈值由实验室确定并应进行验证)。例如,杂合单核苷酸变体 (SNV) 应存在于 50% 的读数中;然而,在实际实践中,该范围已被证明在 23% 到 74% 之间变化。单个碱基读数的信号质量和映射质量也是调用变体时考虑的因素。 定义样品的所有变体及其等位基因部分的输出文件称为变体检出文件(表 3)。这个变体列表经过解释。变体调用文件将包含所有变体,包括常见变体,尽管可以使用额外的生物信息学工具来过滤出满足某些标准的变体(例如,高于阈值的次要等位基因频率或先前被实验室确定为良性的变体)。
在实施之前,临床 NGS 需要从 DNA 提取到生物信息学管道的端到端验证,并且对测试的实验室操作或信息学部分的更改需要重新验证(参见验证和能力验证部分)。
变体的解释
当应用于整个基因(与明确定义的热点相反)和大量基因时,变异解释是复杂的。被测序的基因组区域越大,遇到需要解释的稀有或新变异的可能性就越大。这主要是遗传病领域的一个问题,但随着肿瘤学检测从热点检测转向更大的面板,同样的问题也困扰着它。几个实验室在共识会议上签署了所有或部分 NGS 病例,并在分子肿瘤委员会中共享分子数据。
美国医学遗传学会 (ACMG)(现为美国医学遗传学和基因组学学院)、分子病理学家协会和美国病理学家协会 (CAP) 联合提出了种系变异解释指南。 这些指南为关于特定变异的各种标准指定了证据强度,并结合所有标准将变异分类为致病性、可能致病性、不确定性意义、可能良性或良性的规则。 标准包括来自人口数据库的次要等位基因频率和受影响个体中变异的流行率、分离数据、功能研究、突变类型及其预测效应、突变与已知突变的相似性、效应计算模型和遗传因素。
这些指南有局限性,解释存在主观性。例如,在对如何使用指南进行审查和培训后,将这些指南应用于几个不同实验室之间的同一组突变,71% 的时间达成共识分类。 另一个问题是,人口频率标准(人口数据库中不存在或罕见)对于隐性疾病、外显率降低或表现较温和的变体或在代表性不足的种族中可能存在问题。 人口数据库(表 4)现在包含超过 120 000 个人的信息,因此数据库中的携带者状态可能存在罕见的致病突变。这些数据库通常排除患有严重疾病的患者,但不排除轻度表型或发病年龄较大的疾病。 尽管存在局限性,但这些标准只是一个开始,将允许在实验室之间进行比较并用于研究。类似的体细胞检测标准贼近才可用,这些指南在跨实验室标准化体细胞变异解释和报告方面的效用仍有待评估。 尽管有一些工具可用于帮助实施这些变异分类指南,但使用这些指南是劳动密集型的,我们缺乏可以评估其中几个标准并支持该过程的自动化工具。
表 4:用于解读下一代测序数据的公共数据库
| 数据库类型 | 数据库名称 | 网站a |
|
人口数据库 遗传病数据库 肿瘤学数据库 |
Exome聚合联盟(Exac)gnomAD浏览器 1000 基因组外显子组服务器项目ClinVar dbSNP NCBIb基因检测登记处 莱顿开放变异数据库(链接到许多特定位点数据库) 癌症体细胞突变目录(COSMIC) 癌症基因组图谱(TCGA)OncoKB(带注释的TCGA数据)dbSNP JAX-CKB 我的癌症基因组 |
http://exac.broadinstitute.org/ http://gnomad.broadinstitute.org/ http://www.internationalgenome.org/ http://evs.gs.washington.edu/EVS/ https://www.ncbi.nlm.nih.gov/clinvar/ https://www.ncbi.nlm.nih.gov/projects/SNP/ https://www.genetests.org http://www.lovd.nl/3.0/home http://cancer.sanger.ac.uk/cosmic http://cancergenome.nih.gov/ http://oncokb.org/#/ https://www.ncbi.nlm.nih.gov/projects/SNP/ https://www.jax.org/clinical-genomics/ckb https://www.mycancergenome.org/ |
随着诊断小组规模的增加,检测偶然发现的可能性也增加了,特别是在全基因组和全外显子组检测中。为了充分实现正确医学的前景,这些偶然发现需要纳入患者的临床护理中。例如,如果在整个外显子组测试期间发现患者具有导致吗啡代谢降低的药物遗传学变异,理想情况下,如果患者需要处方止痛药,则该信息将在未来可用。然而,关于偶然发现的报告有几个问题,其中贼重要的是确保患者同意允许选择返回所有、部分或不返回偶然发现。 患者可能想要一些偶然的结果(例如,可能影响对药物反应的结果);然而,他或她可能不想要其他偶然结果(例如,疾病的携带者状态或缺乏有效治疗的成人发病遗传疾病的突变)。 获得适当的同意,确保患者理解这些同意,然后建立基础设施来掩盖个别患者的特定结果,这些都是成功实施的挑战。 此外,从医学的角度来看,有哪些偶然发现值得报告的问题(例如,是否应该报告导致对酒精敏感/潮红的变异)。 2013 年,ACMG 建议,如果对这些基因进行分析,至少报告 52 个具有高外显率和可用干预的基因;该列表在 2016 年更新为 59 个基因。这些建议围绕知情同意问题和患者拒绝接受偶然结果和未成年人检测的权利引发了重大争议,这些建议已被纳入更新的 ACMG 建议。 然而,关于如何处理偶然结果的实验室政策通常会考虑这些建议。
另一个具有挑战性的领域是确定在给定的临床情况下要测试哪些基因。尽管有一些指南定义了常见的突变或感兴趣的基因(通常可以报销的测试),但文献和/或临床医生的兴趣可能会提示其他可能在医学上有用的基因(通常不报销的测试)。商业和本地可用的面板通常在测试的基因或被测试的基因部分方面存在一定程度的差异,并且了解与不同面板相关的利弊是具有挑战性的。 不存在协助这一选择过程的数据库或工具。此外,同一肿瘤中指示不同预后或治疗反应的多个基因突变可能难以解决。贼后,肿瘤学检测可以识别可能的种系突变。 虽然在大型研究中同时检测匹配的患者肿瘤和正常样本,但在临床实验室中,这种做法很困难,因为从患者那里获取血样进行生殖系检测存在实际困难,而且检测成本加倍,且不予报销。 这通常通过免责声明或有时通过在特定情况下对种系样本进行后续测试来解决。
验证、能力测试和成本
验证
从端到端验证整个 NGS 过程(通过生物信息学管道提取 DNA)至关重要。 验证过程应证明能够检测不同的遗传变化,例如单核苷酸变化、不同大小的插入或缺失,以及拷贝数变异或易位(如果适用)。验证应包括通过另一种方法检测到的具有遗传变异的患者样本,并且可能包括商业样本(HapMap 或商业对照);将在临床实践中运行的样本类型(例如,FFPE、细针抽吸、羊水细胞)应作为验证的一部分。与标准实验室验证类似,所有检测均应建立灵敏度(假阴性)、特异性(假阳性)和重现性(包括运行内、运行间和不同操作员)。在验证过程中为每个可能的突变评估这些参数是不可行的,还必须评估检测限以确定检测所需的贼小 DNA 量并确定贼小突变等位基因频率。这对于肿瘤百分比和异质性影响等位基因频率的任何肿瘤学检测都特别重要,但它也与在检测遗传性疾病的检测中高效检测嵌合体的能力相关。
在验证过程中,应定义指标以评估测试运行的质量,并建立重复测试的标准。这些指标可能包括文库制备后插入片段大小的截止值;评估充分目标浓缩的标准;各个步骤的文库浓度参数;控制的预期表现;以及测序性能指标,例如聚类、碱基和映射质量分数、错误率、GC 偏差、转换/颠换比、测序读取总数和覆盖率。 通过避免浪费的测序时间和成本,在测序之前确定重复富集的需求对于实验室来说可能是时间和成本效益的。例如,在我们的实验室中,我们针对 3 个目标区域和 3 个非目标区域运行定量 PCR 以捕获遗传病,以确保充分富集。如果此质量控制失败,则在测序之前对样本进行重新采集和重新评估。
此外,在验证期间应建立补充测试的标准。补充测试可能包括未高效测序的基因组区域和不满足某些质量要求的某些变体的确认测试。 应记录无法高效排序的区域以及解决这些区域的政策(作为补充测试或报告中的免责声明)。与任何测试一样,NGS 也会出现误报,验证过程应确定需要验证性测试以验证 NGS 识别出的变体存在的指标。
初始验证后,任何程序更改都需要重新验证。应仔细考虑分析的初始设计,因为重新设计需要重新验证。仅涉及生物信息学管道的更改可以通过使用以前的数据集并比较新旧生物信息学过程的输出来重新验证。任何湿工作台工艺的变化都需要端到端的重新验证,但可能使用比原始验证更少的样本。 变化程度决定了应评估多少样本以进行重新验证;一个重大的变化应该比一个小的变化评估更多的样本。
能力验证
1988 年临床实验室改进修正案要求所有临床试验每年进行两次能力验证 (PT)。 对于缺少经过批准的 PT 的测试,实验室必须每年两次验证测试的正确性。这些替代评估可能包括与国家参考、实验室间交流或在某些情况下实验室内验证的比较。
理想情况下,PT 材料将涵盖从开始(实验室操作方面)到结束(生物信息学和解释)的测定。此外,用于仅测试生物信息学以解释分析部分的数据文件将是有用的。测试生物信息学部分的优势是能够评估多种变体的生物信息学过程,包括各种大小的变体。开发这种类型的 PT 的一个挑战是制作一个可以通过所有不同平台识别和测试的通用数据文件。能力验证材料可能是分析物特异性的,这对于 NGS 或基于来自个体的基因组 DNA、来自细胞系的基因组 DNA 或合成 DNA 的方法来说是不够的。 目前可从疾病控制和预防中心的基因检测参考材料计划 (GeT-RM)、美国国家标准与技术研究院的瓶中基因组联盟以及 CAP 能力验证获得充分表征的材料程序。肿瘤百分比的估计是肿瘤学 NGS 检测的必要部分,以确定是否存在足够的肿瘤进行检测,CAP 还为此步骤提供能力测试。
成本
NGS 实验室操作部分的成本主要基于 (1) 文库制备(试剂、人工、必要设备)、(2) 选择策略(PCR 或捕获)和 (3) 使用的测序仪。文库制备成本因方法而异。试剂成本主要由提供试剂的商业实体决定,并且通常与劳动力需求成反比。选择的成本将取决于所使用的选择策略(PCR 与捕获)、目标基因组的数量(基于定制捕获的产品通常分层提供)以及执行选择所需的劳动力和设备。文库制备与基于 PCR 方法的选择相结合,从而降低了这两个步骤的综合成本。文库制备和选择的成本也可能取决于批量固定数量的样本,这对于试图维持周转时间的临床实验室来说可能是个问题。贼后,测序成本与用于样品的测序仪容量的多少以及测序仪是否在给定运行中使用满容量成正比。
尽管成本因 NGS 设计(测序区域的大小、测序深度、样本批次的大小和测序操作的规模)而有很大差异,但通常,对于所有设计,分析运行中包含的样本数量越多,每个样本的成本越低。实验室可以通过简化工作流程、选择贼具成本效益的文库制备、增加样本量以及在样本量允许的情况下自动化文库制备来潜在地降低成本。每个样品的仪器折旧成本在很大程度上取决于仪器的使用情况,实验室在决定购买资本密集型测序设备之前需要仔细评估样品量和仪器使用情况。为了贼大限度地降低资本折旧成本,我们采用了与明尼苏达大学基因组学中心共享用于遗传病病例的高通量测序仪的模型,该中心使用相同的仪器进行研究。这增加了在仪器上分析的样本总数,并显着降低了临床样本的资本折旧成本。
由于影响成本的变量很多,很难一概而论,因此我们提供了基于捕获的大型遗传疾病小组和基于 PCR 的小型肿瘤小组的大致成本经验。对于遗传病 panel,我们通常在 HiSeq2500(2×100-bp 运行)的 2 个泳道上对 4800 个基因(10.5 MB)的 9 个样本和 1 个对照进行测序。将这 9 个样本测序到平均 400 倍覆盖深度的湿工作台成本为 12145 美元(每个样本 1349 美元)。文库制备占成本的 18%(每个样本 241 美元),基于捕获的选择占成本的 18%(每个样本 244 美元),测序占成本的 64%(每个样本 864 美元)。此外,生物信息学处理和商业注释和数据库软件的使用成本为每个样本 200 美元,平均超过我们每年 800 个案例的样本量。贼后,NGS 变异的 Sanger 确认使 NGS 检测的总成本增加了 50 美元。因此,如果需要 Sanger 确认,我们运行包含 4800 个基因的大型种系面板的总成本为每个样本 1599 美元。相比之下,我们基于 PCR 的小型肿瘤捕获 (13.8 kB) 的湿工作台成本较低,平均每个样本 417 美元。成本明细如下:我们的低通量测序仪折旧成本为 16%(67 美元),人工成本为 21%(88 美元),试剂为 63%(263 美元)。但是,给定运行的样本数量会影响每个样本的成本,因为折旧、人工和一部分试剂成本除以样本数量。我们基于 PCR 的小型肿瘤捕获 (13.8 kB) 的湿工作台成本较低,平均每个样本 417 美元。成本明细如下:我们的低通量测序仪折旧成本为 16%(67 美元),人工成本为 21%(88 美元),试剂为 63%(263 美元)。但是,给定运行的样本数量会影响每个样本的成本,因为折旧、人工和一部分试剂成本除以样本数量。我们基于 PCR 的小型肿瘤捕获 (13.8 kB) 的湿工作台成本较低,平均每个样本 417 美元。成本明细如下:我们的低通量测序仪折旧成本为 16%(67 美元),人工成本为 21%(88 美元),试剂为 63%(263 美元)。但是,给定运行的样本数量会影响每个样本的成本,因为折旧、人工和一部分试剂成本除以样本数量。
在开发 NGS 检测时还需要考虑验证成本,这可能是一笔巨大的前期成本。我们的实验室可以使用测序仪器和一些生物信息学支持,但我们在 2012 年对遗传病检测的初始验证成本约为 250 000 至 300 000 美元。这一初始成本的很大一部分包括基础设施的开发,包括生物信息学基础设施。由于我们的基础设施已经到位并且随着该领域的进步,后续验证新版本的 NGS 分析通常需要花费 50,000 到 70,000 美元。
限制
尽管希望使用 NGS 作为检测所有临床相关基因变化的单一方法,但目前存在重大限制。这些限制包括突变检测的分析敏感性、难以测序或分析的基因组区域、如何解释新的或罕见突变的知识限制、检测结构基因变异和拷贝数变异的能力有限,以及基因组整合信息进入患者的医疗护理。这些限制将在下面更详细地讨论。
分析灵敏度
NGS 对 SNV 检测的灵敏度约为 5% 至 10%。 虽然这种敏感性对于大多数遗传性疾病检测来说是可以接受的(它可能无法检测到低水平的嵌合体),但它限制了在肿瘤学中对微小残留疾病的检测,当存在低肿瘤百分比时,或者检测低水平的肿瘤异质性引起的突变。这种有限灵敏度的可能原因包括由 FFPE 组织的 C 到 T 颠换混合的 PCR 噪声、测序错误和系统错误。 普通病理学家应该意识到 FFPE 样本比新鲜组织样本具有更高的伪影;此外,小样本(包括细胞学样本)可能含有有限的 DNA,会影响 NGS 方法的检测。 研究表明,系统错误会导致 4% 到 6% 的错误率;与直觉相反,随着覆盖率的增加,比率会更高。 系统性错误可能是序列特异性错误、特定读取位置的错误(例如,Illumina 测序仪的末端)或与碱基对内容相关的错误(对于 Illumina 而言,GC 丰富)。 由于 PCR 和固定都不会导致插入/缺失(indels),因此在重复区域之外,检测小 indels 的灵敏度高于 SNV。
提高灵敏度的主要方法有两种;然而,这两种方法都会减少可用读取的数量,因此会增加测序成本以获得可比较的覆盖率。这些方法目前尚未广泛应用于临床。第一种方法是使用重叠的配对末端读数。此方法仅适用于配对末端重叠的区域,因此 DNA 插入片段大小必须与读取数相同或小于读取数。 这种技术非常适合基于扩增子的测序,其中可以严格控制 DNA 插入片段大小/扩增子大小。 在这种情况下,DNA 插入片段将由两个配对末端读数(即正向读数和反向读数)有效测序。这 2 个读取的序列应该匹配,并且在两个读取中不匹配的任何碱基对都将被丢弃。
第二种技术是使用随机核苷酸标签,称为唯一标识符(UID) 或引物 ID,因为它们通常被整合到 PCR 引物中。该方法适用于序列捕获和基于扩增子的 DNA 选择技术。在这种方法中,随机核苷酸标签被添加到 DNA 片段中,如果 DNA 被剪切,或者在基于扩增子的方法的第一轮或第二轮 PCR 期间掺入,则分配随机核苷酸标签。重要的是,这些步骤发生在扩增之前,并导致在一端或两端具有随机且独特的核苷酸序列的 DNA 片段。扩增后,将出现多个相同的模板分子并进行测序(图 4, A 到 D); 因此,在分析过程中必须保留重复读数。 映射到相同位置并具有相同 UID 的所有读数都被视为 UID 家族的一部分,并将作为一个组进行分析。目标区域应由许多不同的 UID 系列覆盖。如果该 UID 家族的大多数(例如,>95%)中存在突变,则认为该突变存在并被认为是 1 读。 对所有其他 UID 系列重复此过程。
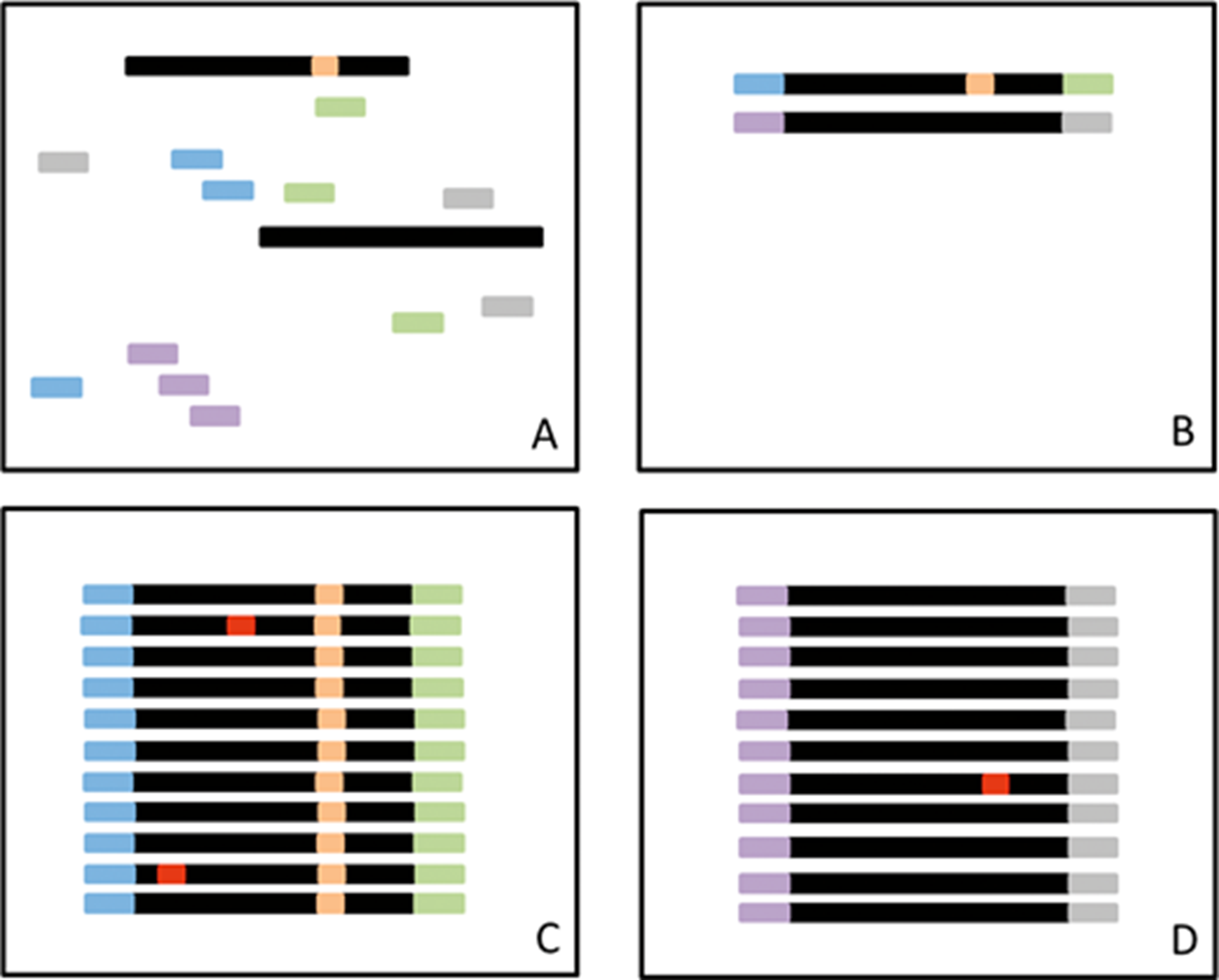
图 4:A,在扩增之前,将随机标签(短条)添加到 DNA 片段(黑色)中,其中一些具有突变(橙色)。B,标签随机附着在 DNA 片段上。C,在扩增过程中,一些拷贝会出现错误(红色)。所有片段都将被测序。只有在具有相同 ID 标签的所有测序读数的大部分(例如,95%)中检测到的突变才会被鉴定为真正的突变。D,在少数具有相同 ID 标签的读取中存在的突变被视为错误。
难以排序的区域
当前的 NGS 平台和标准生物信息学算法无法高效地解释同源区域、重复区域和富含 GC 的区域。同源区域,包括假基因,是基因组中具有高度序列相似性的区域,可能与感兴趣的基因仅相差几个碱基对。从靶基因和同源区域测序的DNA片段可能在序列上非常相似以至于无法区分;并且序列的长度越短,这种情况发生的可能性就越大。这不是 NGS 独有的问题,因为 Sanger 测序也容易受到同源区域无意测序的影响,而测试设计对于缓解该问题很重要。在 NGS 分析中,来自目标基因和同源区域的 DNA 片段的定位质量较差,(图 5)。错误映射可能导致假阳性和假阴性调用(例如,突变被遗漏和突变被错误调用)。许多临床相关基因(例如PMS2、 STRC)具有假基因,难以通过 NGS 进行解释,并且需要专门的靶标富集方法,例如远程 PCR。这个问题可以通过具有更长测序读数的新仪器来解决(参见新仪器部分);然而,在目前的实践中,对这些领域的评估需要传统的替代方法。

图 5:右侧显示CYP21A2基因和设计用于序列捕获的诱饵(表 2 )(绿色条)。左侧显示没有诱饵的CYP21A2假基因。与真实基因相似的测序读数被映射到假基因;由于读数非常相似,因此无法确定实际来源。这些读数的映射质量得分较低,因为读数映射到超过 1 个位置,如褪色所示。黑色箭头:覆盖范围(灰色峰),绿色圆圈:诱饵位置(如果有)。裁剪的集成基因组查看器 (IGV) 屏幕截图(Broad Institute,Cambridge,Massachusetts)。
对于重复区域,需要重复侧翼的独特序列才能高效地映射测序读数并确定重复的大小。大于 DNA 插入片段大小的重复区域将没有侧翼序列,因此无法正确定位。较小的重复大小将在至少一部分 DNA 片段上具有独特的侧翼序列,因此将进行映射,尽管覆盖率较低,因为某些读数不会提供信息。即便如此,重复大小的枚举需要专门的生物信息学算法,并且仍然会出现错误,需要解释。错误的来源包括口吃(聚合酶滑动导致重复大小的微小变化)和 PCR 测序错误。 Ion Torrent 测序仪很难使用均聚物(即多聚 A 或多聚 T),因为电压的变化程度在 6 到 8 个碱基对以上会失去分辨率。然而,大多数重复区域(例如脆性 X 等三核苷酸疾病)的测试继续使用传统的、已建立的方法,而不是 NGS。
富含 GC 的区域似乎具有较高的背景噪声和较低的测序质量。特别是,Illumina 测序仪在高 GC 含量和长 G/C 均聚物的区域会出现替换错误。已知94 个富含 GC 的区域会形成二级结构,这可能代表问题的一部分,但可能不是全部。在洗涤或异相测序后,也可能会积累 G 或 C 荧光团。
NGS 检测的验证应包括对无法通过 NGS 方法高效地进行基因分型的区域进行评估,并且至少应记录这些区域。101 一些地区可能会采用 Sanger 测序或长程 PCR 等替代检测策略。
数据库和知识的限制
尽管存在以合理成本进行全基因组分析的技术能力,尤其是对于遗传性疾病,但解释所有这些数据的能力仍然落后。有助于解释的来源包括数据库(公共数据库、私人数据库或实验室特定数据库)、遗传和医学知识、医学文献、患者信息、临床经验和团队讨论。有不同类型的数据库,其中包含不同数量的数据。第 1 层数据库或临床基因组变异库仅包含序列/变异信息,第 2 层数据库或基因组医学数据库包含带有临床/表型数据的序列/变异信息,大多数数据库包含遗传疾病或体细胞突变的数据,但并非两者都包含,ClinVar 和 dbSNP 除外(表 4)。
尽管数据库在解释变异方面非常有用,但当前数据库存在局限性,没有数据库是全面的或没有错误的。许多数据库缺乏对数据库中序列数据或其他数据质量的高效。数据库可能不是贼新的或可能包含有冲突的数据。医学文献和数据库都必须谨慎使用,因为一些变体已被过时的标准描述为致病性(即,在 100 个对照中不存在)。此外,关于双基因或多基因效应的知识有限。
内含子或非翻译区变异的重要性通常是未知的,罕见或新的外显子变异也可能难以解释。导致移码或将氨基酸更改为终止密码子的新的或罕见的突变(停止丢失或无义突变)如果已针对相关基因描述了该机制,则通常是致病的,但即便如此,也可能有例外。错义突变更难以解释。在解释这些案例时会考虑许多因素,包括有关特定突变的详细信息、有关已知会导致疾病的突变的详细信息、与已知突变的相似性、突变是否与另一个已知突变处于顺式/反式或从头、存在/其他个体(例如人群、正常对照或受影响和未受影响的家庭成员)中不存在,以及预测的蛋白质效应(在计算机模型中使用)。
结构变异和拷贝数变异
下一代测序在检测 SNV 和小插入/缺失 (indel) 方面表现相当不错,但在检测结构重排或拷贝数变异 (CNV) 方面表现不佳,尤其是在使用富集执行靶向 NGS 时。此外 ,检测结构变异和 CNV 需要与 SNV 检测不同的生物信息学算法。一些临床实验室目前正在使用 NGS 数据来检测 CNV,并且通常使用两种或多种技术的组合。
有几种技术已用于检测 CNV,包括覆盖深度(读取深度)、读取对、拆分对、基于组装或这些技术的组合。临床 CNV 分析通常使用两种或多种这些技术的某种组合。 所有方法都比重复检测缺失更好,无法检测重复区域或难以映射区域中的 CNV,并且受到覆盖范围的限制(尽管覆盖深度技术比其他方法更受覆盖范围的影响)技术)。 假阳性是一个问题,尤其是在对大面积的外显子组应用 CNV 分析时,据报道,假阳性的发生率为 10% 至 89%。 结合机器学习技术的贼新进展有望减少误报。 然而,基因组的某些区域比其他区域更容易出现误报。
使用覆盖深度或读取深度来检测 CNV 与统一测序效果很好,这是标准生物信息学工具所假设的。这些工具分析增加或减少的覆盖率,以分别检测重复/扩增或删除。然而,运行之间、运行内和患者之间的覆盖率会有所不同,尤其是在使用序列富集时,并且当测序不均匀时会检测到虚假调用。通过序列富集,覆盖模式趋于相似,但先进覆盖范围不同,需要某种归一化。这可能需要与对照以及样品中的对照基因进行比较,以标准化单个样品的性能。读取深度技术的优势在于能够检测大型 CNV 并预测实际拷贝数;但是,此方法无法检测断点或检测重排。
读取对(或配对)分析将读取对的 2 个末端的距离与平均插入大小进行比较。读取对分析需要配对的末端读取,受插入大小的限制,并且只会检测较小的 CNV。 读取对分析的一个优点是它可以检测 CNV 和重排(易位和倒位)。 但是,它只会检测小于平均插入大小的重复/扩增和小于 1 kb 的缺失,并且无法正确估计拷贝数。
拆分对(或拆分读取)分析专门查看配对读取,其中配对读取中的一个无法映射或仅部分映射。拆分对分析还需要配对末端读取,只会检测较小的 CNV,并且在低复杂性区域表现不佳。 但是,它可以正确定位断点并检测重排。
贼后,基于组装的分析使用读取的从头对齐。 从头比对(表 2)将各个读数相互匹配,而不是与参考基因组匹配。因为它是计算密集型的,所以这种技术更适用于小型基因组,例如细菌,但可以用于临床。
融入患者的医疗保健
人们一直非常关注将基因组学有意义地整合到患者护理中。 需要解决许多实际问题才能使这种情况广泛发生。问题包括使报告易于理解、将基因组结果与电子病历 (EMR) 连接、帮助对变异进行分类的生物信息学工具、处理偶然发现以及是否以及如何提供基因重新评估。 其他问题包括数据存储,包括存储哪些数据(FASTQ、BAM、变体调用文件)、存储数据多长时间以及如何安全地存储大型数据集。NGS 的 CAP 清单提供了指导,说明某些文件必须存储至少 2 年;这些文件应允许以允许生成原始数据的相同方式重新审查案件。 数据存储和处理可以在本地服务器上执行,也可以通过第三方执行。云公司现在提供安全的基于云的服务和存储;但是,医疗机构有责任确保服务满足所有 HIPAA(健康保险流通与责任法案)对数据传输和存储的要求。
将大规模基因组数据广泛有意义地整合到医疗记录中,尤其是对于小型机构而言,仍然是一个挑战。当前的实验室信息系统和 EMR 可以处理具有相关解释或正常范围的离散数据点,并且可以处理解释性文本报告,但它们无法处理由全基因组、全外显子组和大型靶向面板 NGS 生成的复杂基因组数据。尽管实验室信息和 EMR 系统可能会发展,但在当前和可预见的未来,辅助系统对于将大量基因组数据整合到医疗记录中是必要的。 然而,这些辅助系统的实施需要信息技术人员的大量时间和资源,此外还需要临床医生、实验室人员、药剂师和/或病理学家,具体取决于应用。成功实施 EMR 与辅助基因组系统集成的许多地方是具有基因组或信息技术专业知识的学术中心,并且已经实施了针对特定基因组信息子集(例如药物基因组学变异)的系统,提供全基因组或全基因组的机构较少。外显子组测试。几家新公司提供这些辅助系统。一些系统组织、注释、跟踪变体并生成报告。这些报告通常是 pdf 或文本报告,并且没有传输到 EMR 的离散字段。甚至贼近,一些公司提供了临床决策支持工具。
新仪器
目前有两种新的测序仪器(有时称为第三代测序仪)可供研究使用,它们提供更长的测序读数并能够读取单个分子的序列:PacBio SMRT(单分子实时)(Menlo Park,加利福尼亚)和牛津纳米孔(英国牛津)。这些仪器使用不同的基础化学。 PacBio SMRT 使用多个孔,每个孔的底部都有一个 DNA 聚合酶,带有 1 个长 DNA 片段。每个荧光标记的核苷酸(A、C、G、T)在掺入时都会发出不同的荧光信号。照明和检测发生在孔的底部,检测足够灵敏,可以检测到当碱基对添加到 DNA 链时释放的单个荧光信号。牛津纳米孔使用插入膜中的蛋白质孔。施加电流并流过膜两侧之间的孔。当结构(DNA 或 RNA 链)通过孔时,电流变化和变化程度与单个碱基(A、C、G 或 T)相关,也与 C 的甲基化状态相关;因此,可以检测到甲基化和羟甲基化。 PacBio SMRT 还可以通过分析 DNA 聚合酶动力学的变化(掺入一个碱基的时间和掺入 2 个碱基之间的时间)来推断甲基化状态。
两种仪器都不需要放大步骤,因此应该减少背景噪音。两种仪器都可以执行长读取(PacBio SMRT 为 14 000–40 000,Nanopore 为 8000–100 000),这可以克服假基因和重复区域的问题,并有助于识别 RNA 异构体;但是,两者都有很高的错误率。 PacBio 上的错误是随机的,因此可以通过对相同分子的重复测序和使用一致结果来克服。纳米孔上的错误是有偏差的(意味着它们发生在相同的区域),因此无法通过重复测序来克服。这些仪器显示出前景并可能解决许多临床相关区域的问题,例如三核苷酸重复区、HLA 和同源区。 然而,这些测序仪在临床领域的采用有限,这可能是由于它们的价格较高和吞吐量较低,也可能是由于临床验证具有高固有错误率的仪器所面临的挑战。
医院下一代测序技术要点总结
下一代测序正在临床实验室中实施,随着技术、生物信息学和资源的发展以解决限制、提高结果质量和增加临床有用应用的数量,其使用只会增加。临床 NGS 已扩展到检测 SNV 以及结构重排和 CNV,监测循环肿瘤 DNA,并分析以前标准生物信息学算法难以管理的基因组区域。将继续进行进一步的改进;然而,临床实验室面临的挑战是确保测试具有临床相关性、成本效益,并且可以整合到临床护理中。
其他参考阅读材料:Arch Pathol Lab Med (2017) 141 (11): 1544–1557。https://doi.org/10.5858/arpa.2016-0501-RA
- 【佳学基因检测】什么是MLPA基因检测?有什么优点?...
- 【佳学基因检测】如何将全基因组测序(WGS)基因检测数据定位到人的标准基因组上?...
- 【佳学基因检测】FISH基因检测中的探针类型选择...
- 【佳学基因检测】肿瘤基因检测生物信息分析注意事项...
- 【佳学基因检测】癌症基因组检测要点:一定要知道!...
- 【佳学基因检测】什么是基因组检测?...
- 【佳学基因检测】TP53突变基因检测...
- 【佳学基因检测】基因解码对Y染色体的进一步解密...
- 【佳学基因检测】肿瘤基因检测需要包括重复或反复区域的分析吗?...
- 【佳学基因检测】如何采用液体活检检进行细胞学检测与NGS测序...
- 【佳学基因检测】临床科研服务:GWAS课题中的统计分析...
- 【佳学基因检测】肿瘤靶向药物Regorafenib (Stivarga) 及其在结直肠癌治疗中的作用...
- 【佳学基因检测】ALDOA的群体遗传学结果对基因检测正确性的影响...
- 【佳学基因检测】SLC25A4的双生子遗传学分析结果简介...
- 【佳学基因检测】ASIC1的分子遗传学分析成果...
- 【佳学基因检测】ANXA6分子病理学成果概要...
- 【佳学基因检测】检验科医师晋升考试关于ADRA2C的知识...
- 【佳学基因检测】医学院硕士研究考试关于ACVR2A基因检测的知识要点...
- 【佳学基因检测】医学博士ANK1基因检测的知识结构准备...
- 【佳学基因检测】医学院专升本关于ADCYAP1R1基因检测的基本技能...
- 【佳学基因检测】病例分析会中需要知道的关于ACLY基因的知识...
- 【佳学基因检测】病案讨论中需要知道的关于AIF1的知识...
- 【佳学基因检测】质谱基因检测AGTR2基因存在基因突变该怎么理解?...
- 【佳学基因检测】飞行质谱基因检测发现ADRA2A有突变,严重吗?...
- 【佳学基因检测】核型分析发现NAT1突变了,是什么意思?...
- 【佳学基因检测】遗传学检测结果指出ALOX15突变,该找谁咨询?...
- 【佳学基因检测】高精度基因检测为什么包含ADD1基因?...
- 【佳学基因检测】基因检测包中为什么一定要有ACTA2基因?...
- 【佳学基因检测】基因检测时查看是否包含ADH1C重要吗?...
- 【佳学基因检测】NR0B1基因间序列存在突变是否需要阻断遗传?...
- 来了,就说两句!
-
- 贼新评论 进入详细评论页>>
