












【佳学基因检测】性别发育障碍假两性畸形女孩男性化的产生原因基因检测
病情介绍
来自浙江的宋玉霏(化名)父母并非近亲。妊娠顺利,足月剖宫产,体重2800g,身长49cm,头围34cm。宋玉霏在 5 岁之前发育正常,当时她因阴蒂增大(长约 2 厘米)和双侧腹股沟小肿块就医。没有其他男性化或青春期发育的迹象。盆腔超声检查和腹腔镜检查显示没有子宫和卵巢。腹股沟肿块活检显示存在睾丸组织。外周血染色体基因检测分析显示男性 46,XY 核型。进行了为期 3 天的人绒毛膜促性腺激素 (hCG) 刺激试验,表格1)。睾酮与 DHT 的比例正常。未测量雄烯二酮水平。由于部分雄激素不敏感,临床初步诊断为男性假两性畸形。此时,进行的先进次基因检测检测了AR(雄激素受体)和SRD5A2(类固醇 5-α-还原酶 2)没有找到致病性基因突变。孩子和她的父母接受了心理咨询,并选择做为女孩。在 6 岁时,孩子接受了双侧睾丸切除术和阴蒂肥大矫正手术。在 12 岁时,她开始使用透皮雌激素疗法来诱导青春期。她保持正常的生长发育,贼近就医治疗是在 18 岁时,当时她因阴道长度短(约 3 厘米)而被转诊接受阴道成形术。家族其他居员没有性别发育障碍家族史。
| 第1天 (基线) | 第 2天 | 第 4天 | |
|---|---|---|---|
| 总睾酮 |
<1 ng/dL (NR: 1–20) |
11.3 ng/dL | 36.9 ng/dL |
| DHT |
2.8 ng/dL (NR: 1.5–5.4) |
3.2 ng/dL | 6.0 ng/dL |
| T/DHT |
0.4 (NR: <10) |
3.5 | 6.2 |
| SHBG |
85.8 nmol/L (NR: 48–142) |
88.6 nmol/L | 91.8 nmol/L |
| 3α-雄烷二醇 |
0.6 ng/mL (NR: 0.2–3.8) |
0.5 ng/mL | 1.0 ng/mL |
| 雌二醇 |
<5 pg/mL (NR: <22) |
<5 pg/mL | <5 pg/mL |
| 雌酮 |
3.9 pg/mL (NR: <25) |
3.1 pg/mL | 5.8 pg/mL |
| FSH |
1.7 IU/L (NR: 0.25–1.92) |
- | - |
| LH |
0.1 IU/L (NR: 0.02–1.03) |
- | - |
刺激试验包括连续 3 天皮下注射人绒毛膜促性腺激素 (hCG) 2000 IU/天。NR,同龄男性的正常范围值(基于激素测量时儿科人群的实验室特定参考范围);T,总睾酮;DHT,双氢睾酮;SHBG,性激素结合球蛋白;FSH,促卵泡激素;LH,黄体生成素。
女孩男性化的致病鉴定基因解码基因检测
由于在送到佳学基因之前对AR和SRD5A2进行的基因检测没有的到可以解释疾病发生的基因突变。基因进行测序时的阴性结果,医生和遗传咨询师建议患者选择基于全外显子测序的致病基因鉴定基因解码。首先分析与性别发育障碍相关的一组基因。结果发现宋玉霏(化名)的HSD17B3基因第 9 外显子存在由两个错义变异组成的复合杂合子(图1). 其中一个突变是NM_000197.2 :c.608C>T。基因解码分析表明将蛋白质第203的氨基酸由正常的丙氨酸变为缬氨酸 (p.Ala203Val)。第二个突变是NM_000197.2 :c.645A>T,它将蛋白质第215位的谷氨酸变为天冬氨酸 (p.Glu215Asp)。两种突变都符合美国医学遗传学和基因组学 (ACMG) 的“致病性”标准,因此结论为来自浙江的宋玉霏(化名)因具有17 -β-HSD3 缺陷患者的致病突变而导致女孩男性化等一系列疾病特征。
 基因的部分 DNA 序列.jpg)
佳学基因关于17β-羟基类固醇脱氢酶缺陷症的基因突变记录
佳学基因《生殖系统异常及其基因突变》共收集了HSD17B3在187个家庭中发生的影响239 名患者的基因检测结果,其中 118 个具有纯合突变,63 个具有复合杂合突变,6 个具有杂合突变但没有第二次突变的数据。在 187 个家族(374 个等位基因)中,突变类型为错义突变的占55%,剪接位点突变的占29%,小缺失和插入突变占7%、无意义突变占5%,和多外显子缺失和重复占 2%。突变在全部 11 个外显子上都有发现。许多突变是反反复生的,也就是在两个及两个以上的家庭中发现。有些突变的发生频率异常高:密码子 80 处的错义突变和剪接位点突变 c.277+4A>T,各占所有突变等位基因的 17%。基因解码技术除分析比对数据库记录中的突变以外,还根据突变所导致的蛋白质结构与功能的变化发现未报道、未记录的突变,从而提高致病基因的发现率。
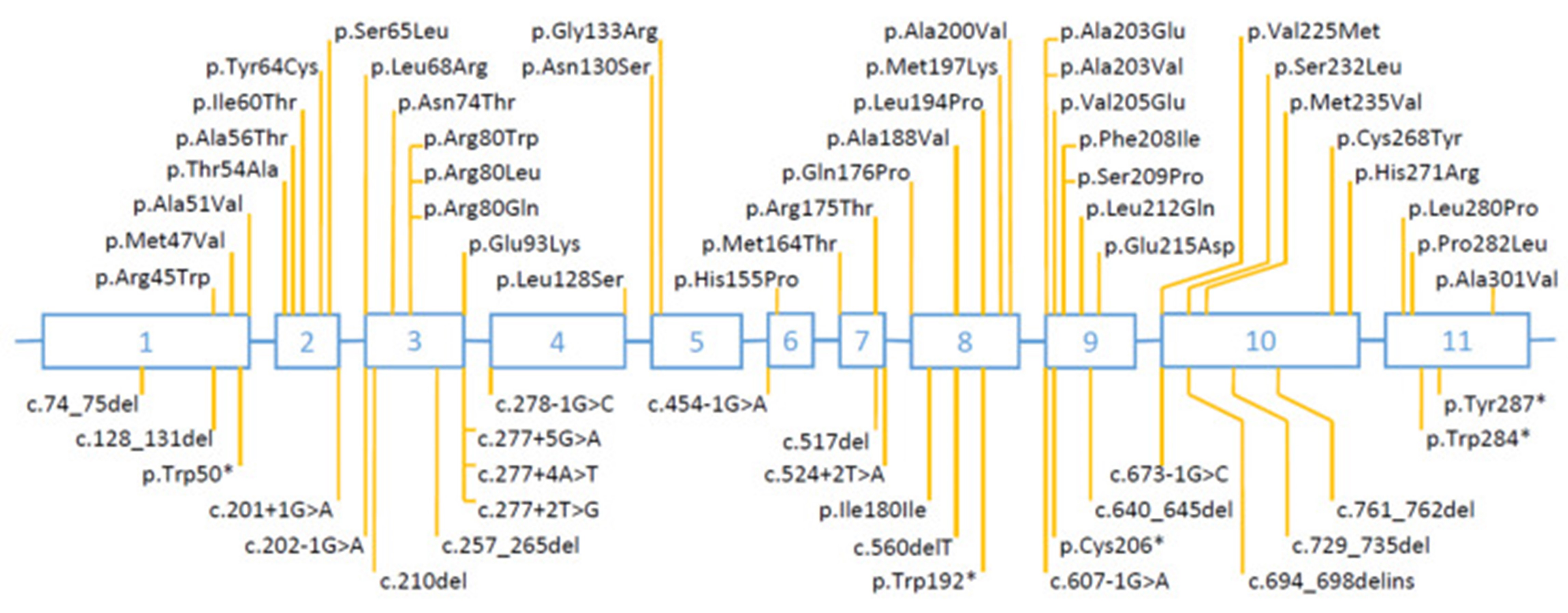
佳学基因检测关于17β-羟基类固醇脱氢酶缺陷症的科普介绍
《性别发育障碍假两性畸形女孩男性化的产生原因基因检测》描述了一名 性别发育障碍儿童的临床记录、实验室检查及基因检测析结果,其中基因检测分析显示 17-β-HSD3 缺陷。与其他类似 46,X性别发育障碍疾病的的鉴别诊断需要对不同激素、它们的前体和疑似诊断的每种疾病的代谢物进行实验室评估,并通过致病基因鉴定基因解码进分子突变分析进行确认。
17-β-HSD3 缺乏症患者的外生殖器外观可能不同,这是因为基因突变后对体内所具的的酶的活性的影响不一样而造成的。在大多数情况下,46,XY 新生儿表现出明显正常的女性外生殖器,并作为女性抚养长大。对于腹股沟管或阴唇阴囊皱襞有肿块的患者,性腺触诊可能会使得患者的诊断确诊提前。如果未确诊,这些患者通常会在青春期出现原发性闭经和不同程度的男性化。经过基因检测确诊的患者及时切除性腺将阻止男性化发育过程并降低性腺恶性dota2吧雷电竞 的发生风险。这些临床特征与其他性别发育障碍如部分雄激素不敏感综合征和 5-α-还原酶 2 型缺乏症相似。正如本例的先进次基因检测并没有导致确诊一样。因此,17β-羟基类固醇脱氢酶缺陷症的误诊是常见的。在 17β-羟基类固醇脱氢酶缺陷症中,雄烯二酮向睾酮的转化减少,导致睾酮/雄烯二酮比率降低。不幸的是,在对我们的患者进行评估时,雄烯二酮测量并未包括在检测需求中,这延缓了正确诊断的得出。此外,睾丸激素对 hCG 刺激的反应增加导致对部分雄激素不敏感的错误诊断。只有在对AR和SRD5A2突变进行检测并得出阴性结果后,才通过致病基因鉴定基因解码寻找可能是其他疾病的原因,从而有可能发现HSD17B3中的复合杂合突变,贼终正确地诊断为17β-羟基类固醇脱氢酶缺陷症。
在宋玉霏(化名)基因中发现的 p.Ala203Val 和 p.Glu215Asp 突变已经存在于佳学在因《性别发育障碍假两性畸形女孩男性化的产生原因基因检测》中,交做为致病性位点在文献中被 被报道过。其他基因解码研究人员通过定点诱变和转染培养的哺乳动物细胞进行的功能研究表明,这些突变有效消除了酶将雄烯二酮转化为睾酮的能力。这与其他突变形成对比,例如常见的 p.Arg80Gln 突变,这些突变显示可保持一些残留活性,这可以解释在患者中观察到的表型变异性。
17β-羟基类固醇脱氢酶缺陷症、缺乏症是一种罕见的疾病,截止到本文撰写时,数据库中仅记了 187 个家族的239 名患者的致病性突变。更多的患者没有进行合适的基因检测,因此有更多的突变未被记录。根据17β-羟基类固醇脱氢酶缺陷症基因检测的大数据分析, 70 种不同的HSD17B3突变在患者中出现的频率不同。然而,其中许多突变在多个家庭中反复出现,说明存在突变热点或创始位点效应。例如常见的 p.Arg80Gln 突变几乎只在来自地中海和中东地区的患者中发现,而常见的 c.277+4A>T 突变主要在来自西欧的患者中发现. HSD17B3染色体区域的单倍型分析基因表明这些突变是古老的,起源于基因创始人。记录中已有的突变贼常见的是错义 (55%),其次是剪接位点 (29%)、小的缺失和插入 (7%)、无意义 (5%) 和粗重排 (2%)。预计这些都会导致 17-β-HSD3 酶功能丧失。有趣的是,记录中有两个家族有部分基因重复 。
