
2017年4月,患儿,男性,6岁,因“突发意识不清伴抽风1 h”于2014-02-15入院。病史采集:0.5 d前(入院当日)突然出现意识不清、呼之不应、全身无力症状,1 h前(就诊途中)发生惊厥,表现为牙关紧闭、口吐泡沫、双眼凝视、颈项强直、四肢抽搐,急诊考虑为“惊厥待诊”给予降温、镇静处理后收入院。家族史:患儿祖父有色素脱失斑史,惊厥史不详,患儿父亲有皮肤色素脱失斑及面部血管纤维瘤,有惊厥发作史。既往史:生后即发现皮肤色素脱失斑,婴幼儿期惊厥发作数次,智力发育落后于同龄儿,情绪易激动,受责备时有抓打头部等自伤行为。3周岁至今尚无惊厥发作。
体格检查:体温36.5 ℃,心率100次/min,呼吸27次/min,血压96/64 mmHg(1 mmHg=0.133 kPa),体重18 kg。身材矮小,营养不良,意识清楚。无皮疹,颈部触及数个黄豆大小可活动淋巴结,质中。咽(-),双肺呼吸音粗,可闻及少许粗湿啰音。心(-)。腹平坦,腹壁静脉曲张显露,右下腹可见两个手术瘢痕,无压痛、反跳痛。肝肋下4 cm,质中,边锐,无触痛,脾肋下1 cm,质软,边锐,无触痛。肠鸣音正常,双侧睾丸未有效降入阴囊。脊柱四肢活动自如,无畸形,可见杵状指(趾),下肢无浮肿。四肢肌力、肌张力无异常,克、布氏征阴性,双侧巴氏征(-)。
进一步检查:血常规、C反应蛋白及ESR正常;大便常规见脂肪颗粒;肝功能不同程度升高:丙氨酸氨基转移酶71.8 U/L,天冬氨酸氨基转移酶118.0 U/L,总胆汁酸205.5 μmol/L,碱性磷酸酶394 U/L,r-谷氨酰转肽酶169 U/L,其余未见异常;血脂降低:胆固醇2.3 mmol/L,高密度脂蛋白0.44 mmol/L,低密度脂蛋白1.21 mmol/L;免疫球蛋白IgG、IgA、IgM、IgE未见异常,类风湿因子(-),T/B细胞亚群未见异常,抗核抗体、抗双链DNA、抗Sm抗体均阴性、血糖、微量元素及凝血功能未见异常,乙型及丙性肝炎抗体(-),梅毒及HIV检测(-);多次痰及肺泡灌洗液培养为铜绿假单胞菌,余病原学检查无阳性发现,心脏彩超正常,腹部B超示肝脏弥漫性纤维化。进一步腹部增强CT提示怀疑肝间质纤维化,肝硬化、脾大、门脉高压、侧支形成;门静脉肝内分支变细;胰腺显示欠佳。住院期间多次行支气管镜检见鼻咽部大量黄白色脓性分泌物,鼻窦口可吸出大量黄白色黏性分泌物,气管、左右主支气管及各叶段支气管管腔内均可见大量黄白色脓性分泌物。完善囊性纤维化跨膜传导调节蛋白(CFTR)基因检测,发现患儿CFTR基因c.648G存在纯合突变(图12),并检测患儿父母CFTR基因,均为c.648G>A的杂合子(图13)。考虑患儿诊断为囊性纤维化(cystic fibrosis,CF)。
在某基因检测公司囊性纤维化全套基因检测没有发现基因变化。经医生推荐选择佳学基因致病基因鉴定基因解码。
佳学基因取患儿3ml抗凝静脉血,采用致病基因鉴定基因解码技术发现患儿TSC2基因C.481+5G>T存在杂合突变,该病为显性遗传。双亲中未以生突变,属患儿在发育过程中的新发突变。该突变在千人数据库、dbSNP数据库、ESP6500基因数据库均未记载。
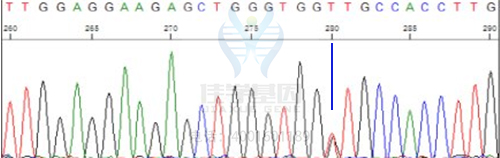
患儿所含有的一个突变基因,未在父母中发现,是个体发育中的新发突变,可自然生育第二胎。
导致该患者罹患疾病的基因突变位点不存在于任何国际、国内数据库中,包括千人数据库、dbSNP数据库、ESP6500基因数据库等数据库均不包含此位点。根据国际数据库设计的基因检测和采用数据库比对方法所进行的基因解读分析无法找到并明确患者的致病基因。需要采用致病基因鉴定基因解码的方法解读独特基因序列的病理影响才能找到致病基因。
结节性硬化症(Tuberous sclerosis complex,TSC)又称Bourneville病,是一种常染色体显性遗传的神经皮肤综合征,发病率约为1/6000-10000。是一种由基因的序列变化引起的基因病、遗传病。基因变化序列部分遗传自父母,部分是由个体的发育过程中自发产生的。临床常见特征是面部皮脂腺瘤、癫痫发作和智能减退。表现为多器官受累,主要累及源于外胚层组织的器官,如神经系统、皮肤和眼等。中枢神经系统肿瘤是该病致死及致残率的更主要原因,病理改变表现为神经胶质增生性硬化结节,90%患者出现室管膜下胶质结节,70%有皮质或皮质下结节。6-14%患者可出现室膜管下巨细胞星型细胞瘤(SEGA),这种肿瘤可阻塞脊液通路致脑积水和颅内压增高。神经系统损害为本病更常见严重症状,主要表现为1、癫痫:发病率约占70-90%,自婴幼儿期开始,发作样式多样,可自痉挛症开始,至部分性局灶性或反复性发作、性大发作。2、智能减退:多进行性加重。癫痫发作伴高峰节律异常脑电图者常有严重的智能障碍,部分患者可表现为孤独症。3、少数可有神经系统阳性体征,如锥体外系体征或单瘫、偏瘫、截瘫、腱反射亢进等。Roach等2004年建议的诊断标准为目前临床诊断TSC的指导标准。但仍需要同其他多系统疾病进行鉴别诊断。根据基因定位可将结节硬化症分为二型:TSC1,TSC2。TSC1和TSC2突变分别引起错构瘤蛋白(hamartin)和结节蛋白(tuberin)功能异常,影响其细胞分化调节功能,从而导致各个胚层细胞生长和分化异常。遗传方式为常染色体显性遗传,家族性病例约占三分之一,即由父母一方遗传而来突变的TSC1或TSC2基因;散发病例约占三分之二,即出生时患者携带新突变的TSC1或TSC2基因,并无家族成员患病。家族性患者TSC1基因突变较为多见,而散发性患者TSC2基因突变较为常见。
本例患儿具有皮肤色素脱失斑、反复惊厥症状和明确家族史,TSC较为典型但未引起重视,院外多次被误诊为“高热惊厥”。这提示对该类患儿应注意详细采集病史及其家族史,并仔细进行体格检查及采取必要辅助检查以尽早明确诊断。部分TSC患儿以惊厥为先进症状就诊,被误诊为“癫痫”,后经影像学检查确诊为TSC。目前TSC无特异性治疗方法,常采用对症治疗,如控制癫痫发作,婴儿痉挛可采用促肾上腺皮质激素(ACTH)治疗,并应用脱水剂降低颅内压,若脑脊液循环受阻则可手术治疗,对面部皮脂腺瘤可行整容术治疗。此外,本例患儿日常生活出现性格精神改变,在受到家长责备时有抓打头部等自伤行为,提示对该类患儿应注意心理疏导。经佳学基因解码后,患者可优先采用国际上正在研发的新型治疗药物,同时进入基因矫正优先治疗序列。
佳学基因致病基因鉴定基因解码可以分析发现三万多种疾病发生的基因原因。联系电话:4001601189。佳学基因官网://www.looklottery.vip。以下为佳学基因检测分析的疾病种类的少量举例:

招商热线:010-52802095 邮箱:jiyinjiema@163.com 邮编:102206
地址:北京市昌平区中关村生命科学园生命园路8号 网络支持: 基因解码基因检测信息技术部
