












【佳学基因检测】dota2雷竞技 基因检测明确 3 型 Usher 综合征家庭的突变位点
眼科基因检测导读:
Usher综合征(USH)是一种严重的遗传性疾病,主要表现为听力和视力的双重丧失。USH最早由苏格兰眼科医生Charles Usher描述。据估计,先天性耳聋患者中USH的发病率约为3.5/100,000至16.6/100,000。该病是先天性重度或深度耳聋儿童的根本病因之一,占大约9.2%的病例。此外,USH仅次于Pendred综合征,是最主要的综合征性耳聋形式。
USH的主要临床表现包括听力丧失、视网膜色素变性、前庭功能障碍等。不同类型的USH在表现和发病时间上各有差异。USH为常染色体隐性遗传。至今,眼科疾病的致病基因鉴定基因解码鉴定并确认了9种致病基因,分别为:Usher 1型(USH1)的MYO7A、USH1C、CDH23、PCDH15和SANS;Usher 2型(USH2)的USH2A、ADGRV1和WHRN;Usher 3型(USH3)的CLRN1。
进行性听力损失是USH3的显著临床特征。USH3患者通常在8-10岁时被诊断出听力损失,但在20至30岁之间会出现快速进展的听力丧失。视网膜色素变性(RP)通常在青春期后被诊断,平均诊断年龄为17岁,夜盲症是最早出现的症状。与听力损失的快速进展不同,USH3患者的视网膜色素变性的进展与USH1和USH2患者的进展没有显著差异。青春期后,视杆细胞退化,导致进行性视野丧失,最终发展为管状视野和失明。此外,USH3A患者还表现出不同程度的前庭功能障碍,约51%的受检患者在前庭测试中显示异常。
早在1995年,Eeva-Marja Sankila等人将唯一负责USH3的基因定位于3q21-25位点。随后,2002年,Avital Adato等人在人类和小鼠中鉴定了USH3A转录本。通过分析USH3A的表达模式,利用原位杂交技术在小鼠的耳蜗毛细胞和螺旋神经节细胞中鉴定出该基因的转录本,并将其命名为clarin-1。经过近十年的努力,人类CLRN1基因的位点最终被确定为3q25.1。通过基因解码明确,与其他四跨膜蛋白类似,CLRN1仅保留有限的亲水区,受胞质或胞外水相影响,明显缺乏任何功能结构域,至少已有三种剪接异构体被报道(见图1)。所有已知的CLRN1突变均位于异构体a的三个外显子上,除了异构体e的外显子0和外显子0b之间的内含子区域内的内含子突变。基因解码表明,CLRN1基因的主要异构体编码一个232个氨基酸的蛋白质,预测该蛋白质具有四个跨膜结构域和一个位于第一个细胞外环中的糖基化位点。
CLRN1在小鼠耳蜗毛细胞毛束的形态形成和维持中起着重要作用,也可能具有突触作用。体外生化分析表明,CLRN1可作为分子支架发挥作用,在不同质膜区域募集参与细胞粘附的蛋白质,并在组织肌动蛋白细胞骨架的组织中发挥作用。与该功能一致,Clrn1敲除(KO)小鼠和N48K敲除小鼠在幼年时表现出发育不良的毛束,并在出生后第21天(P21)严重失聪[。然而,与其他USH疾病小鼠模型类似,这些小鼠并不表现出USH3患者的眼部表型。尽管在临床患者中存在遗传和表型特征,但CLRN1的分子功能及其与其他Usher基因产物的关系和相互作用通过数据库比对基因检测难以明确。
佳学基因眼科疾病基因检测病案集描述了一个遗传性耳聋家系的分子病因学基因解码。该家系共有2名患者,跨越3代。所有患者均出现双侧神经性听力损失(SHL)、进行性视力丧失和夜盲症。通过详细的临床病史资料,结合完整的体格检查、影像学检查和分子病因学评估,评估永生化淋巴细胞系CLRN1基因的分子致病机制。本案例明确家系的特殊临床表型和分子病因,为家系及后代提供实用有效的遗传咨询,并为USH3在中国人群的长期基因解码提供更多资源。
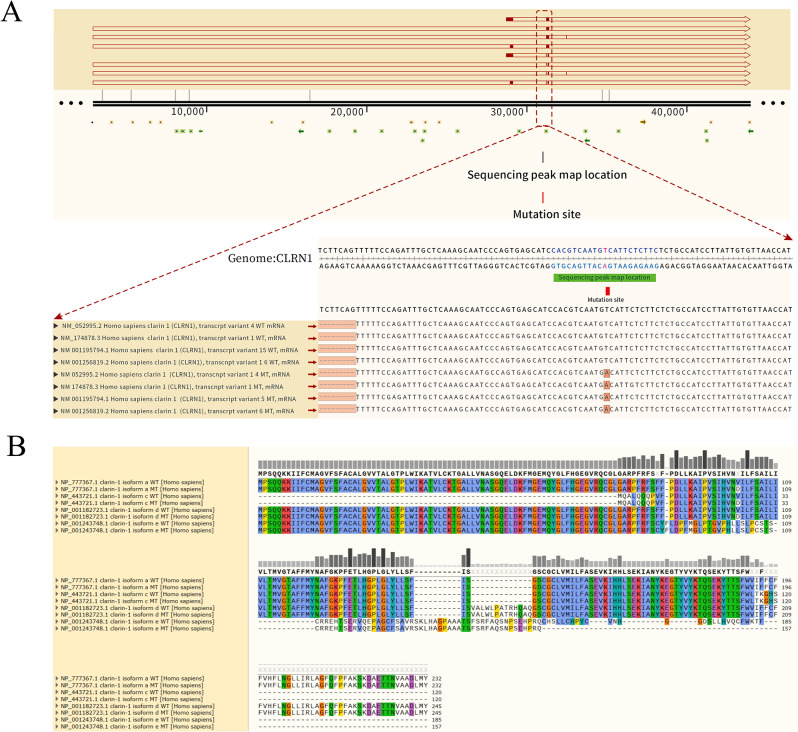
(A)突变位点出现在 CLRN1 的不同转录本中。(B) CLRN1 的点突变可导致不同转录本中出现不同的氨基酸序列变化
临床评估
临床评估由耳鼻喉科医师、眼科医生和临床遗传学家完成,包括耳镜检查、视觉强化、听力检查、鼓室测量、声反射、纯音听力检查或播放听力检查、失真产物诱发耳声发射 (DPOAE)、听觉脑干反应 (ABR) 和听觉稳态反应 (ASSR)。在 0.25–8.0 kHz 的倍频程下确定双侧气导 (AC) 阈值。测量 0.5、1、2 和 4 kHz 对话频率下的 AC 平均阈值,并用于定义听力损失严重程度。听力水平被标记为轻微 (16–25 dB)、轻度 (26–40 dB)、中度 (41–70 dB)、重度 (71–95 dB) 或极重度 (95 dB)。对部分家属进行最佳矫正视力、双眼视觉诱发电位(VEP)、视野及眼底检查,并进行高分辨率计算机断层扫描(HRCT)检查,以核实家属是否患有听力障碍以外的并发症。
外周血样本
在获得知情同意后,从所有家系成员、500 名正常对照个体(263 名男性和 237 名女性,年龄 18 至 25 岁)和 122 个无关中国家系采集血液样本(2 ml + 5 ml)。
靶向基因捕获和高通量测序
采用苯酚/氯仿法从受试者的外周血白细胞中获取基因组DNA。使用Nanodrop 2000分光光度计对DNA进行定量。采用下一代测序(NGS)技术鉴定该家系的致病基因。在靶向捕获和大规模并行测序(MPS)过程中,使用Covaris S2聚焦超声(Covaris,马萨诸塞州,美国)随机切割符合一定标准的先证者(II-4)的合格gDNA,平均片段大小为350–400 bp。然后修复片段尖端,连接到适配器,并使用Agilent 2100生物分析仪进行分析。使用GenCap试剂盒(MyGenostics,北京,中国)捕获所有外显子和侧翼内含子区域,6个与耳聋相关的线粒体区域和139个与耳聋相关的核基因的3个miRNA(补充 表1、2和3 )。使用 NextSeq 500 下一代测序仪(美国加利福尼亚州圣地亚哥 Illumina Inc.)对捕获的序列进行高通量测序。
临床表型分析
患者来自某三代遗传性耳聋家系(见图2A),该家系为常染色体隐性遗传。先证者为Ⅱ-4,女性,47岁,其父母Ⅰ-1和Ⅰ-2为近亲结婚。患者双耳听力严重减退(图2C),双眼视力低下。大约在10岁时,患者开始出现双耳听力逐渐丧失的情况;13岁时,视力障碍逐步加重,并伴有夜盲症。过去10年内,双眼视力明显下降。
眼科检查(如图2E所示)显示,左眼的最佳矫正视力(BCVA)为0.4,右眼为0.7。双眼的视觉诱发电位(VEP)检查结果显示,双眼P100波峰的潜伏期延迟,且幅度减小。双眼视野检查结果提示出现管状视野。双眼彩色眼底检查结果表明,视盘边界清晰,呈蜡黄色;黄斑区反光不清;视网膜血管变窄;视网膜呈灰白色,赤道部视网膜可见色素沉着,这些都是晚期视网膜色素变性(RP)的典型表现。

家谱图(A)、突变信息(B)、听力图(C)、颞叶CT扫描(D)和眼科信息(E)
(A)家族家谱图:填充的黑色符号(男性为方形,女性为圆形)表示患病个体(II-1、II-4),空心符号代表未患病个体。箭头指向患儿。
(B)DNA序列色谱图:显示II-1和II-4的CLRN1基因突变,分别为c.474T > A(P.Cys158Ter,NM_001256819.2)和c.302T > A(p.Val101Asp,NM_174878.3)这两种新型纯合变异。
(C)听力图:患病兄弟姐妹的听力图显示双侧严重神经性耳聋。横轴表示音频频率(Hz),纵轴表示听力阈值(dB)。
(D)颞叶CT扫描:颞叶CT扫描结果未见明显的异常。
(E)眼科检查:双眼VEP检查显示患儿双眼P100波峰潜伏期延迟且波幅减小。双眼视野检查结果提示双眼出现管状视野。双眼彩色眼底检查显示,双眼视神经乳头边界清晰,呈蜡黄色;黄斑区反光不清;视网膜血管变窄,视网膜呈灰白色,赤道部视网膜出现色素沉着,这些表现均为晚期视网膜色素变性(RP)的典型特征。
II-1为先天性耳聋患者,出生后听力反应不佳,自幼视力不佳,但未做过眼科检查,已丧失视力及生活自理能力。
靶向高通量测序及共分离验证结果
佳学基因对先证者 (II-4) 的 139 个耳聋基因的所有编码外显子加上约 100 bp 的侧翼内含子序列进行了测序。目标区域的平均覆盖深度在 110–160 倍之间,其中 > 98% 的覆盖深度 > 20 倍。因此,佳学基因从面板分析中获得的结果不仅在很大程度上排除了 Usher 综合征基因和导致类似综合征的基因编码序列中的突变,例如 ABHD12 突变和HARS突变,而且还排除了与耳聋和 RP 相关的基因同时发生的突变,导致模仿 Usher 综合征的表型。最后,在转录本NM_001256819.2处检测到一种新的纯合变体,其中致病性并不总是被报告,CLRN1:c.474T > A (P.Cys158Ter),在患者 II-4 和 II-1 中均被检测到。如果变体为 c.302T > A,则基于 MANE 选择转录本NM_174878.3,该转录本为 p.Val101Asp。
p.Cys158Ter 突变影响 Clarin-1 功能
CLRN1 c.302T > A ( NM_174878.3 )突变导致101位缬氨酸残基变为天冬氨酸,如图 5所示,Alphafold2预测软件显示CLRN1 c.302T > A导致alpha折叠异常,并未引起其他空间结构变化。而CLRN1 c.474T > A ( NM_001256819.2 )突变发生在第158位氨基酸,对应cDNA产生终止密码子,导致翻译提前终止。大片段氨基酸的丢失最终影响蛋白质的空间结构(图 6 ) 。CLRN1 c.474T > A ( NM_001256819.2 )致病性的预测结果见(表 6、7)。
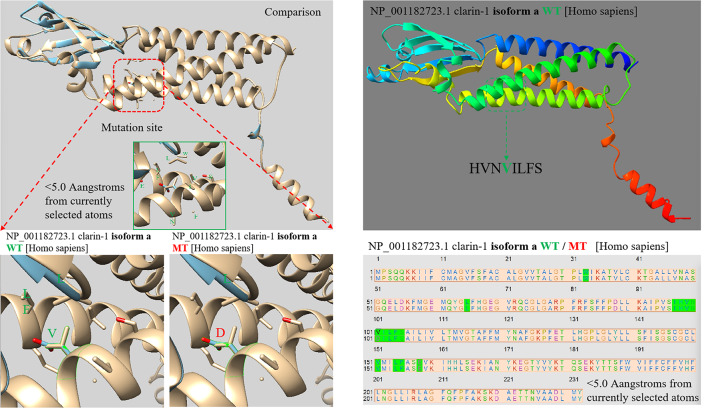
Alphafold2 预测软件显示 CLRN1c.302T > A 导致 alpha 折叠异常,并未引起其他空间结构变化
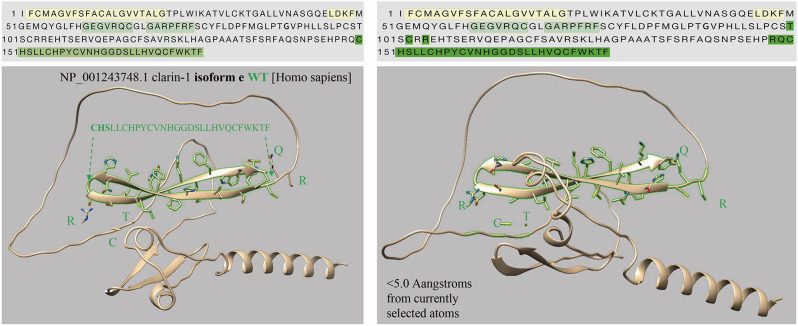
CLRN1 c.474T > A( NM_001256819.2 )突变发生在第158个氨基酸处,相应cDNA产生终止密码子,导致翻译提前终止。
基因检测解读
本案例对一个跨三代遗传性耳聋家系中的2名患者进行了临床表型和分子病因学分析。根据先证者和家系中部分患者表现出的进行性听力丧失与视力丧失,并结合先证者的眼科检查,初步诊断为USH3。经分子检测,发现这2名患者均携带CLRN1基因的纯合突变:c.474T > A(P.Cys158Ter,NM_001256819.2)或c.302T > A(p.Val101Asp,NM_174878.3)。随后,采用Sanger测序验证了这些变异位点,并证实了CLRN1 c.474T>A(P.Cys158Ter,NM_001256819.2)或c.302T>A(p.Val101Asp,NM_174878.3)突变与该家系的基因型-表型共分离。
USH3的临床表现主要为语言后进行性双侧对称性神经性耳聋(SHL),伴随前庭功能减退,通常会出现视网膜色素变性(RP),且发病年龄一般在20岁之前[7]。尽管如此,USH3的临床表型具有显著的遗传异质性和表型特异性。目前已知,CLRN1基因突变是USH3A的主要致病原因,而HARS基因突变则与USH3B相关[15]。与USH1和USH2相比,USH3较为罕见,尽管Hope等[16]曾报道USH3占伯明翰地区USH患者的约20%,而Ness等[5]则指出,该亚型在芬兰和阿什肯纳兹犹太人群体中约占40%。然而,在西班牙人群的基因解码中,对89名无USH3病史的USH患者进行分析时,只有8例为USH3患者,发病率仅为6%[8]。在中国人群中,尚未有关于家族性USH3的报道,现有病例多为散发的眼部疾病相关病例。
由于USH3患者的表型异质性不仅在不同家系之间存在显著差异,甚至在同一家系内也表现出较大的异质性,Ness等[5]认为,合并SHL的USH3患者在表型上较为一致,可作为区别USH3与USH2、USH1的重要标志。另一个常用的区分标准是耳聋的发病时间,语后SHL是USH3的典型特征,而语前耳聋则更常见于USH1和USH2患者。
在本案例中,先证者展现了典型的USH3临床症状,尤其是在听力、视力以及典型的RP方面,其发病年龄和进展与文献报道一致。然而,她的哥哥(II-1)则表现出不同的临床表型:从出生开始双耳即为重度SHL,夜盲症早期出现,且双眼视功能较差,几乎完全丧失。因此,家族内USH3患者的临床表现存在一定差异。Adato等[17]曾报道,在一例也门犹太裔非近亲家族中,出现了两种不同的临床表型:一例为USH1,另一例为USH3,表明在同一家系内,USH的临床表型可能因不同突变类型而有所不同。类似地,Ness等[5]也在德系犹太人群中发现,虽然所有USH患者均携带CLRN1基因突变,但携带p.N48K纯合突变的患者表现出更为严重的临床表型,尤其在听力损失的早期和快速进展上有明显特点。
尽管USH3的听力损失进展较为缓慢,但一些病例与USH2A、USH1B及USH1D的变异表现相似。随着病例数据的积累,USH3的临床表型已逐渐呈现更多样化,基因诊断的准确性和可靠性也得到提高。基于本案例和其他文献的分析,进行性SHL和语后耳聋并不一定能作为诊断USH3的决定性标准。
本案例的重要意义体现在以下几个方面:首先,NCBI网站记录了CLRN1基因的四个转录本(图1),由于这些转录本包含的外显子不同,因此它们编码的氨基酸序列也有所差异,这有助于解释为何CLRN1突变会导致临床表型的多样性。NM_174878.3和NP_777367(isoform a由转录变体1编码)包含外显子0-1-2,编码一个约25.8kDa、含232个氨基酸的clarin-1蛋白[18]。已知的CLRN1突变,除了位于isoform e的外显子0和外显子0b之间的内含子区域外,其他都位于isoform a的外显子3上。除芬兰人群中的p.Y176X和p.P120K突变[19],以及阿什肯纳兹犹太人群中的p.N48K突变[20]外,其他多数突变均在单个家族中发现。除这些常见突变外,迄今已报道了21种错义突变、无义突变、2种异常剪接突变、7种缺失突变和3种插入突变(截至2023年3月的人类基因突变数据库)。
2017年,Khan等[21]报道了一个CLRN1内含子突变c.254–649T > G(NM_001256819.1,异构体e),该突变发生在阿拉伯半岛的一个近交系USH1患者中,位于外显子0和外显子0b之间的内含子区域。实验显示,该突变导致外显子剪接异常,进而导致移码和提前终止密码子的产生。这一突变也被认为是一个创始等位基因,对阿拉伯半岛的聋盲患者有显著影响。
通过使用Alphafold2预测软件,我们发现家族性患者携带的CLRN1突变[CLRN1 c.474T > A(P.Cys158Ter, NM_001256819.2)或c.302T > A(p.Val101Asp, NM_174878.3)]对不同转录本的影响各异。虽然我们未能从患者来源的ILCL中获得直接证据,但在HEK293T细胞中,我们确认了特定转录本的表达,并通过质粒表达实验观察到c.474T > A(P.Cys158Ter, NM_001256819.2)引起的蛋白表达差异。这一机制可能是由于突变导致的可变剪接,从而引起蛋白质降解。
CLRN1 c.474T > A(P.Cys158Ter)变异尚未见报道,至今未在任何公共数据库(如单核苷酸多态性数据库、人类基因突变数据库等)中出现过。根据ACMG/AMP致病性分析指南,这一变异通过体外实验验证,已被证明会导致蛋白表达异常并损害基因功能(PS3),且在正常人群中未见该变异(PM2)。结合临床表现和家族基因型-表型共分离的证据(PP4和PP1),我们最终判断CLRN1 c.474T > A(P.Cys158Ter)为致病突变,并认为这是中国人群中首次报道的USH3突变。
基因检测结论
本案例确定CLRN1 c.474T > A纯合变异(P.Cys158Ter,NM_001256819.2)在中国家系中引发USH3。该突变与芬兰和阿什肯纳兹犹太人群中的常见版本不同,因此其发现为USH3A的发病机制和预防基因解码提供了新的视角,也提示我们在基因解码不同转录本时应特别关注其可能的临床影响。
(责任编辑:佳学基因)
