












【佳学基因检测】多囊肾病基因检测:常染色体隐性多囊肾病
多囊肾病之常染色体隐性多囊肾病基因检测
常染色体隐性多囊肾病 (ARPKD) 是一种罕见疾病,也是多囊肾病最严重的形式之一,可导致儿童期终末期肾病 (ESRD)。PKHD1是导致绝大多数 ARPKD 的基因。然而,一些病例与最近发现的新基因(DZIP1L基因)以及几种可以模仿 ARPKD 样表型谱的纤毛基因有关。此外,使用细胞和动物模型阐明了与 ARPKD 发病机制和进展有关的许多分子通路。然而,ARPKD 蛋白的功能和疾病的分子机制目前仍未完全了解。在这里,我们回顾了 ARPKD 的临床、治疗、遗传学和分子基础,重点介绍了该领域的最新发现。
多囊肾病基因检测:常染色体隐性多囊肾病关键词
ARPKD、囊肿、罕见单基因疾病、肾脏病学
为什么要做多囊肾病基因检测?
常染色体隐性多囊肾病 (ARPKD) 是一种严重的遗传性囊性疾病,以双侧肾囊性疾病和先天性肝纤维化相结合为特征。ARPKD 表现为围产期或儿童期症状,是儿童发病和死亡的重要原因。ARPKD 是一种罕见疾病;在芬兰的一个孤立近交群体中,每 8000 个新生儿中就有 1 个患有该病。然而,在中国,报告的发病率为每 26485 个新生儿中就有 1 个,年患病率为每 100,000 人中有 1.17 人。ARPKD 的普遍患病率估计为每 20000 个新生儿中就有 1 个。
ARPKD 是一组异质性单基因疾病的隐性形式,称为多囊肾病 (PKD)。显性形式常染色体显性多囊肾病 (ADPKD) 具有更高的流行病学患病率,通常在成人中诊断。
什么样的人就当做常染色体隐性多囊肾病基因检测?
ARPKD 的表型高度多变,可表现为围产期、新生儿、婴儿、青少年或青年期发病的疾病,且没有已知的性别或种族偏见。通常情况下,最严重的 ARPKD 病例出现在妊娠晚期或新生儿期,双侧肾脏肿大且回声增强,皮髓质分化差,肾形轮廓保留,并有多个微小囊肿。此外,他们还可以表现为羊水过少或羊水不足,导致典型的“Potter 序列”表型,有肺发育不全、特征性面部特征和马蹄足挛缩的肢体。除 Potter 序列外,其他肾外表现并不常见。目前尚无记录表明存在肝脏表型,但已报道了一些相关表型,如腹腔难产。
由于严重羊水过少伴有肺发育不全的风险,因此与更糟糕的预后相关。多达 50% 的 ARPKD 新生儿死于肺发育不全和胸腔压迫导致的呼吸功能不全。然而,围生期后,存活率很高,1 年和 10 年存活率分别达到 85% 和 82% 。围生期存活下来的患者需要内科专家的精心护理。请注意,产前诊断和终止妊娠是该疾病流行病学中需要考虑的因素。除了早期尿毒症和肺未成熟之外,肾脏肿大和肺发育不全还会导致另一个问题,即肠内喂养困难,这可能会使营养复杂化,需要持续鼻胃管喂养。
在大多数情况下,ARPKD 在生命最初几个月内的严重问题是动脉高血压,需要使用多种药物干预治疗。严格控制血压对于防止高血压造成进一步的肾脏损害至关重要。但肾衰竭并不是新生儿死亡的常见原因 。肾脏疾病的表现包括尿路感染、肉眼血尿以及儿童早期的肾性骨病。关于超声异常,通常在肾脏肿大之前报告回声增强和肾囊肿。肾脏肿大是由于远端肾单位广泛扩张,通常发生在集合管中。随着病情的进展,肾脏结构逐渐类似于 ADPKD 中的模式,出现不同大小和外观的肾脏大囊肿,常伴有间质纤维化。这种情况导致生命最初几十年内几乎 50% 的患者出现终末期肾病 (ESRD),需要进行肾脏替代治疗。
尽管该病被称为“常染色体隐性多囊肾病”,但囊性肝表型在该病中起着重要作用,这也解释了为什么原发基因被称为“多囊肾和肝脏疾病 1 ( PKHD1 )”,其中多囊肝病 (PLD) 是主要的肾外表现。这些组织学变化是肝导管板发育缺陷的结果,称为导管板畸形 (DPM) [ 23 , 24 ]。DPM 也是其他纤毛病的常见特征 [ 4 ]。随着患者年龄的增长,会出现肝脏疾病。最早的表现是先天性肝纤维化 (CHF),肝内和肝外胆管均有不同程度的扩张(Caroli 综合征)[ 23 ]。在 ARPKD 病程中,肝脏疾病有两种主要表现:由于进行性肝纤维化导致的门静脉高压;和胆管炎 [ 14 ]。门脉高压症相关的并发症包括脾肿大、血小板减少和食管静脉曲张,可产生严重的出血并发症 [ 14 , 25 ]。成年 ARPKD 患者(40 岁以上)与罹患肝脏dota2吧雷电竞 (尤其是胆管癌)的风险之间存在一定的联系 [ 24 ]。值得注意的是,肝细胞功能通常保持稳定,血清肝酶在正常范围内,胆汁淤积参数除外 [ 4 , 12 , 23 , 26 ]。这一事实使目前的临床方法难以监测肝病的严重程度和进展 [ 25 ]。
尽管约 10% 的 ADPKD 患者会发生脑动脉瘤,但 ARPKD 中仅描述了少数病例 [ 27 ]。颅内动脉瘤的最高危险因素是高血压,ADPKD 和 ARPKD 患者都有这种疾病 [ 28 ]。一些动脉瘤在早期就被诊断出来,这一点很引人注目,因为儿童动脉瘤非常罕见 [ 27 ]。此外,ARPKD 患者还可出现其他肾外和肝外表现,包括左心室肥大、反复呼吸道感染、神经系统异常、眼底异常、腹痛、脓毒症发作以及脊柱和四肢畸形 [ 14 , 29 ]。
虽然大多数 ARPKD 患者的病情发展相似,但表型并不典型;在老年人群中,一些 ARPKD 病例报告为肝脏或肾脏中度受累,甚至为唯一或主要的表型 [ 4 , 21 ]。这与以下事实有关:尽管 ARPKD 是一种隐性疾病,杂合子携带者不应表现出任何临床疾病表现,但数据表明PKHD1突变杂合子患 PLD 和轻度 PKD 的风险增加 [ 30 , 31 ]。
去:
3.诊断
由于 ARPKD 症状早期且严重,因此通常在产前就被诊断出来。在产前诊断中,从妊娠中期/晚期开始的超声检查可以检测到肾脏肿大、回声增强以及髓质回声增强,这是由于皮髓质分化消失、肝实质回声增强且伴有纤维组织。羊水过少会使诊断更加困难,因此需要进行超声检查和 MRI 检查。30% 的 ARPKD 病例报告发现微囊肿(5-7 毫米),但大囊肿(>10 毫米)很少见,可能提示患有其他不同的纤毛病。这些超声发现在其他病理中很常见,例如梅克尔综合征,产前超声可能无法检测到该疾病的轻度形式。在这些情况下,基因检测可以提供准确的诊断 [ 32 , 33 ]。
鉴定出PKHD1基因后,便可以通过直接 DNA 测序(桑格法)进行基因诊断。然而,由于基因组规模庞大以及疾病相关突变的等位基因异质性,PKHD1突变的基因检测十分复杂 [ 34 ]。然而,根据遗传学工作组的建议,在疑似 ARPKD 病例中应避免进行单基因检测,因为其表型谱重叠广泛。作为替代方案,下一代测序等方法已成为人们感兴趣的技术,因为它们可以在独特的测试中同时有效地分析多个候选基因,而且成本相对较低。在极少数情况下,甚至可以在具有严重新生儿临床表型的儿童中观察到两个基因的突变 [ 4 , 33 , 35 ]。
基因检测的结果对于每种疾病相关的合并症和并发症的临床管理至关重要,可以提供明智的遗传咨询,并在未来实现更有针对性的精准医疗 [ 4 ]。
去:
4. 鉴别诊断
ARPKD 表型不仅是由PKHD1基因突变引起的。这使得诊断和管理(包括围产期护理)变得困难。还需要考虑许多其他隐性和显性基因(图1) [ 4,21,36 ] 。
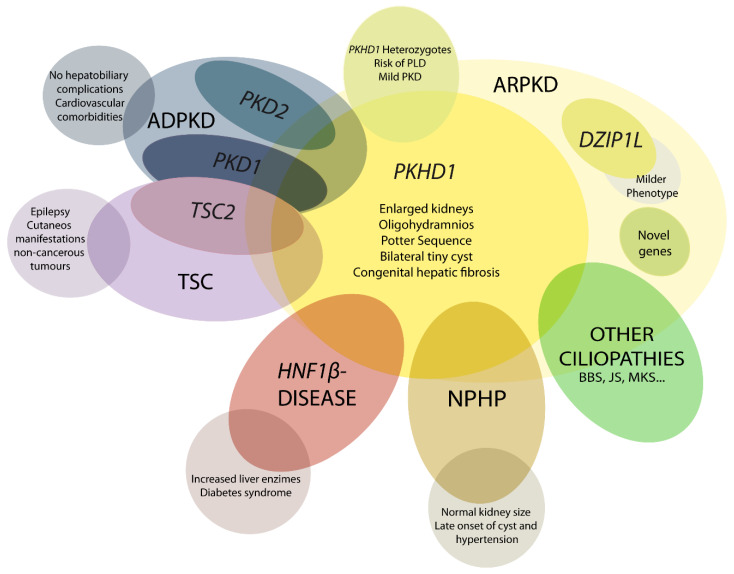
图1
ARPKD 鉴别诊断示意图。PHKD1是 ARPKD 的主要致病基因,而DZIP1L表现出较轻的表现型。其他基因突变可能与 ARPKD 的临床表现重叠,例如PKD1和PKD2是常染色体显性多囊肾病 (ADPKD) 的主要致病基因;TSC2 导致结节性硬化症 (TSC);以及其他例如HNF1β、肾痨 (NPHP) 基因和其他纤毛病,如 Bardet–Biedl (BBS)、Joubert (JS) 和梅克尔综合征 (MKS)。这些重叠的表现型表明纤毛病基因之间存在生理复杂和功能相互作用。
4.1. DZIP1L 相关多囊肾病
在携带DZIP1L突变的患者中,ARPKD 的中度表现已被描述[ 37 ]。与该基因相关的首例报告病例发生在产前或幼儿期;然而,尚未报道围产期死亡病例 [ 4 , 21 , 37 ]。
4.2. 早期重度常染色体显性多囊肾病(ADPKD)
在大多数情况下,ADPKD 不会在成年之前出现临床症状,但也有一小部分病例(最多 5%)在儿童期或出生前就表现出疾病。目前尚无确定的理由认为随机、表观遗传和环境因素会改变表型。如果一个孩子患有早发性 ADPKD,那么其他兄弟姐妹中重复发病率通常很高,这让我们相信存在家族性修饰因素,例如复杂的遗传相互作用与第二个修饰因素。“剂量敏感网络”可能会使细胞完整性恶化,并可能解释这些患者更严重的临床病程 [ 4 , 21 , 38 , 39 ]。
临床上,ARPKD 与 ADPKD 有显著的重叠,但每种疾病也都有一些特征性表现。与 ARPKD 患者不同,ADPKD 患者很少出现肝胆并发症。此外,ADPKD 患者更容易出现心血管合并症,尤其是颅内动脉瘤 [ 21 ]。
4.3. TSC2-PKD1 导致的早期和严重 PKD
TSC1和TSC2基因突变会导致结节性硬化症 (TSC)。这种多器官疾病主要与癫痫和皮肤表现有关。患者可能会在肾脏、心脏和脑中出现非癌性肿瘤。肾脏表现是成年患者死亡的主要原因 [ 40,41 ]。当 16p 染色体上的缺失影响两个相邻基因(PKD1和TSC2 )时,通常会导致 PKD 早期发作。此外,它们之间的密切生理相互关系可以解释 PKD 和TSC之间的临床重叠;TSC2蛋白功能已被证明在协助多囊蛋白 1 定位方面发挥作用 [ 21 ]。
4.4. HNF1β相关疾病
HNF1β/TCF2基因突变会导致产前肾脏回声增强和波特氏序列,以及可能与 ARPKD 混淆的多囊肾肿大。将这些相似表型联系起来的一种解释是,除了其他多囊肾基因外, HNF1β还是调节PKHD1表达的转录因子。然而,该基因突变可导致多种表现:肾囊肿和糖尿病综合征;生殖道缺陷;内分泌/外分泌腺疾病、低镁血症和肝酶升高 [ 21 , 42 , 43 ]。
4.5. 肾痨(NPHP)
NPHP 被归类为常染色体隐性囊性肾病,其特征是伴有纤维化的肾小管间质囊肿。迄今为止,大约有 20 个基因与隐性 NPHP 有关 [ 36 ]。与 ARPKD 不同,NPHP 肾脏仍然很小。囊肿和高血压通常仅在晚期疾病中表现出来,这是 25 岁以下患者 ESRD 的主要原因之一。在某些情况下,表现可能类似于 ARPKD,肾脏肿大或 Potter 序列。NPHP 蛋白与其他纤毛病蛋白在功能网络中发挥作用;NPHP 基因的鉴定和特性有助于了解囊肿形成的分子机制 [ 4 , 21 , 44 , 45 ]。
髓质囊性肾病(最近被命名为小管间质肾病 TKD)是由MUC1和UMOD突变引起的,被认为是常染色体显性 NPHP,其发病时间比隐性形式晚 [ 21 ]。
4.6. 其他纤毛基因突变
基因突变可能与 ARPKD 相似,而这些基因通常会导致其他(通常更复杂)纤毛病,例如 Bardet–Biedl (BBS)、Joubert (JS) 和 Meckel 综合征 (MKS)。BBS 表型可能不均一,但通常表现为肾脏肿大和回声增强,并伴有皮髓质分化丧失。最严重的纤毛病是 MKS 和 JS,其特征是早发性发育障碍和神经系统问题,以及许多纤毛病的特征,例如肝纤维化、多指畸形和囊性肾 [ 4 , 21 ]。
另一个例子是 Von Hippel–Lindau,这是一种由肿瘤抑制基因 VHL 基因突变引起的常染色体显性遗传病。这会导致中枢神经系统血管母细胞瘤,并伴有肾脏肿瘤。患者也有很大概率出现肾脏和胰腺囊肿。TSC、VHL 和 PKD 表现之间的相似性表明存在功能联系:初级纤毛和 mTOR 信号通路 [ 4 , 46 ]。
去:
5. ARPKD的遗传学
正如我们之前提到的,ARPKD 是由PKHD1 基因突变以及最近发现的DZIP1L 基因突变引起的[ 37 , 47 , 48 ]。PKHD1位于6 号染色体 (6p12.3-p12.2) (图 2PKHD1基因是人类最大的基因之一,其基因组片段约为500 kb。预测该基因至少有 86 个外显子,以复杂的可变剪接变体模式组装,转录出约 8.5 kb–13 kb 的大型全长 mRNA [ 49 ] 。已在该基因中鉴定出多种被认为是致病的突变类型。目前,已鉴定出约 750 个PKHD1突变,其中约一半是错义突变。外显子 3 中的错义突变 (c. 107C>T; p.Thr36Me) 是最常见的突变,占所有病例的 20% 以上 [ 34 , 51 ]。这种突变是在杂合子中观察到的,具有第二个不同的突变等位基因 [ 29 ]。大多数病例是家族性的,但也有报道称存在新生突变,占 2% 至 5% 的病例 [ 34 ]。有趣的是,在孤立性常染色体显性多囊肝病 (ADPLD) 的背景下,Besse 等人报道了几例PKHD1突变的杂合子携带者,他们队列中的 102 名 ADPLD 患者中有 10 名是由PKHD1突变导致的,其中一名出现 p.Thr36Me 错义变异 [ 30 ]。根据临床观察,10% 的 ARPKD 患者出现无数无症状肝囊肿是一个遗传事实 [ 31 ]。然而,这些数据不足以解释为什么ARPKD 中的PKHD1会导致严重的肝肾表型,而在 ADPLD 中却不会;在这方面,还需要更多的研究 [ 52 ]。
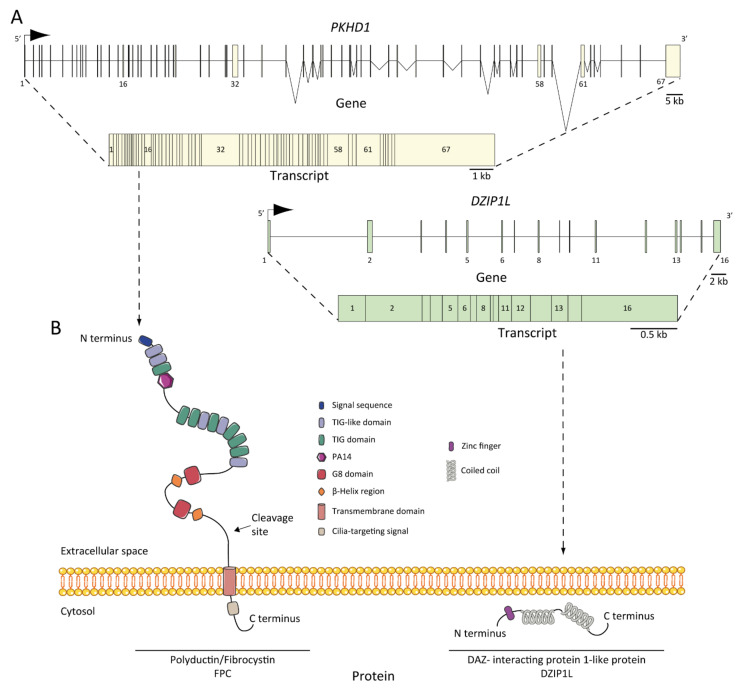
图 2
ARPKD 基因、转录本和蛋白质:( A ) PKHD1和DZIP1L基因和转录本。外显子的位置已说明并编号,最长的转录本来自两者:PKHD1有 67 个外显子, DZIP1L有 16 个外显子;( B ) 纤维囊蛋白/多聚导管蛋白 (FPC) 和 DAZ 相互作用蛋白 1 样蛋白 (DZIP1L) 的结构。蛋白质未按比例绘制。
最近,Lu 等人通过全基因组 SNP 分析、全外显子组测序和 Sanger 测序 [ 37 ] 建立了位于 3 号染色体 (3q22.1-q23) 上的DZIP1L (图 2A),编码 767 个氨基酸的蛋白质,作为与 ARPKD 发病机制相关的新基因。作者在七名具有 ARPKD 表型的患者(约占其队列的 0.3%)中发现了突变,而没有证据表明 PKD 基因中存在另一种突变。他们在两个家族中发现了与该疾病分离的纯合错义突变(p.Gln91His 和 p.Ala90Val),并在另外两个不相关的近亲家系中发现了纯合蛋白质截断突变(p.Gln155* 和 p.Glu354Alafs*39)。数据表明,DZIP1L突变不是该疾病的常见原因,但尽管如此,ARPKD NGS 诊断多基因组应该针对该基因,原因有二:DZIP1L 突变可能与其他 PKD 或纤毛病基因座相互作用,并有助于拓宽对该疾病遗传复杂性的理解 [ 37 , 53 ]。
基因型-表型相关性
复合杂合子使得建立可能的基因型/表型相关性变得复杂。最常见的突变 c.107C>T (T36M) 仅能解释约 20% 的突变等位基因,但其他突变均未被描述为一个簇;事实上,许多变异都是单一谱系所独有的。PKHD1 中存在大量致病变异,包括截短、错义和内含子/剪接突变。通常,基因型-表型研究更多地针对突变类型,而非突变的具体位点 [ 54 , 55 ]。
携带两个截短突变的患者表现出严重的表型,围产期或新生儿死亡率高,而存在一个或两个错义突变的患者通常表现出中等表型。然而,也有例外,Ebner 及其同事描述的一名携带两个PKHD1截短突变的患者在没有肾脏替代疗法的情况下存活了生命的前 30 个月,或者相反,几例携带两个错义突变的患者可能与截短变体一样严重 [ 55,56,57,58 ]。
此外,多达 20% 的兄弟姐妹表现出明显的表型差异,这意味着基因型不足以解释 ARPKD 的表型变异,其中复杂的转录谱可能发挥重要作用 [ 54,59 ]。此外,有证据表明,在具有其他基因变异的 ARPKD 小鼠模型中,该疾病得到了改变;例如,Pkhd1和Pkd1突变的共同遗传会使表型恶化;这与人类中另一个 ADPKD 基因(PKD2)的突变也会使肾脏表现恶化有关 [ 60,61 ] 。人们还认为,表观遗传学和环境因素,以及与PKD相关的其他基因的遗传变异,可以解释家庭间和家庭内的变异。
去:
6. ARPKD蛋白:结构和功能
PKHD1的蛋白质产物是纤维囊蛋白/多聚导管蛋白/FPC(图 2B) [ 34 , 48 ],是一种膜蛋白,具有较长的胞外 N 末端、一个跨膜结构域和一个短的胞质 C 末端尾巴。胞外结构域含有 12 个 TIG/IPT 结构域(Ig 样结构域),这些结构域已在细胞表面受体中描述 [ 62 ]。此外,在胞质尾中还鉴定了三个潜在的蛋白激酶 A (PKA) 磷酸化位点,可能与其功能有关 [ 63 ]。据报道,PKHD1同源物PKHD1L的同一性为 25%,相似性为 41.5%,它编码纤维囊蛋白-L,这是一种具有诱导性 T 淋巴细胞表达的受体,与 PKD 无关 [ 64 ]。FPC 的最长开放阅读框 (ORF) 预测为 4074 个氨基酸长 [ 65 ]。然而,PKHD1基因经历了复杂的剪接模式,并能编码多种额外的基因产物。同样,FPC 表现出高度复杂的 Notch 样蛋白水解加工模式,该模式已在体外水平 [ 66 ] 和使用小鼠模型的体内水平 [ 67 ] 得到验证,这使得对PKHD1 /FPC的研究特别困难。
FPC 是一种 440 kDa 的膜结合蛋白,主要在肾脏(皮质管和髓管)、肝脏(肝内和肝外胆管)和胰腺(胰管)中表达 [ 65 , 68 , 69 ]。检测到了两种分别为 ~230 和 ~140 kDa 的 FPC 产物,更重要的是,在分泌的 FPC 产物的细胞级分中发现了 ~140 kDa 产物 [ 65 ]。在亚细胞水平上,FPC 在肾上皮细胞和胆管细胞 [ 71 ] 的初级顶端纤毛 [ 65 , 68 , 70 ] 和纤毛基底体区 [ 69 ] 中表达。此外,FPC 也在集合管细胞的顶端膜和细胞质中表达 [ 65 ]。 ARPKD组织是否缺乏FPC表达还存在争议,一些研究支持这一观点[ 48,70 ],但其他证据则表明并非如此[ 72 ],这表明FPC剪接变体在时间和空间上表达的复杂性。
FPC 的结构表明细胞表面受体可能具有功能,它通过 N 端与细胞外配体相互作用,或通过 C 端将细胞内信号传导至细胞核 [ 73 ]。胞质尾部在全长裂解后可转位至细胞核 [ 66 , 74 ]。然而,C 端的内在机制仍不清楚,因为在小鼠模型中,C 端的缺失不会导致肾脏或肝脏囊性表型,这表明它对 ARPKD 中的囊肿形成并非至关重要 [ 67 ]。
DZIP1L编码 DAZ(无精子症缺失)相互作用蛋白 1 样,这是一种锌指蛋白,具有多个卷曲螺旋结构域和一个 N 端附近的 C2H2 型锌指结构域 [ 37 ]。锌指蛋白 DZIP1L 参与初级纤毛的形成 [ 75 ],Lu 及其同事认为它可能在多囊蛋白/PC(ADPKD 蛋白)运输中发挥作用 [ 37 ]。研究结果强调纤毛过渡区是研究 ARPKD 发病机制的一个新的潜在关键点 [ 53 ]。
去:
7. ARPKD的发病机制/分子基础/发病机理
7.1. ARPKD 啮齿动物模型:从动物模型中吸取的教训
到目前为止,已经开发出几种与人类 ARPKD 非常相似的动物模型(表格1PKD 的早期模型是由非直系同源基因的自发突变引起的,这些基因模仿了该疾病的隐性性状和表型 [ 76 ] 。1985年报道的第一个小鼠模型是先天性多囊肾小鼠,或cpk [ 77 ]。人们对该模型中疾病的发展、表现/外显率以及遗传学进行了广泛的研究 [ 76 , 78 ]。cpk模型会导致 C57BL/6J (B6) 品系发生自发突变,该突变与Cys1基因相对应 [ 62 ]。后来,在 20 世纪 90 年代,出现了其他基因座自发突变的模型,包括已充分研究的pcy小鼠 [ 79 ],其Nphp3基因座发生突变[ 80 ]。此外,还描述并表征了BALB/c 多囊肾 ( bpk ) [ 81 ] 和幼年囊性肾模型 ( jck ) [ 82 ],它们分别在Bicc1 [ 83 ] 和Nek8 [ 84 ] 中发生自发突变。随后是通过化学诱导设计的动物模型,如使用苯丁酸氮芥诱变程序获得的幼年先天性多囊肾 ( jcpk ) [ 85 ]。有趣的是,后来的研究表明,bpk和jcpk模型改进了Bicc1基因中的突变位点[ 83,86 ]。此外,通过插入诱变,从大规模插入诱变程序中发现了Oak Ridge 多囊肾或orpk小鼠 [ 87,88 ] 。
表格1
ARPKD 的当前动物模型列表,或模仿其表型的动物模型列表。根据已发表的数据,动物模型按从最旧到最新的顺序排列。
| 型号名称 | 硬币 | 基因 | 等位基因类型(突变类型) | 肝脏表型 | 肾脏表型 | 其他表型 | 参考。 |
|---|---|---|---|---|---|---|---|
| 模拟ARPKD的动物模型 | |||||||
|
cpk ( Cys1 cpk ) * |
老鼠 | 半胱氨酸1 | 自发的 |
∙肝囊肿 ∙胆管扩张 ∙肝纤维化 |
∙肾囊肿 ∙肾脏肿大 |
∙胰腺囊肿 | [ 77,93,94,95,96 ] |
|
pcy ( DBA/2-pcy/pcy ) ( Nphp3 pcy ) * |
老鼠 | Nphp3 | 自发的 | 没有任何 |
∙肾囊肿 ∙肾脏肿大 ∙肾纤维化 |
∙颅内 动脉瘤 |
[ 79,80,97 ] |
|
bpk ( Bicc1 jcpk-bpk ) * |
老鼠 | Bicc1 | 自发的 | ∙胆管扩张 |
∙肾囊肿 ∙肾脏肿大 |
∙过早死亡 ∙出生后死亡 |
[ 81 ] |
|
jck (Nek8 jck)* |
老鼠 | NEK8号 | 自发的 | ∙无 |
∙肾囊肿 ∙肾脏肿大 |
∙过早死亡 | [ 82 ] |
|
orpk ( Ift88 Tg737Rpw ) * |
老鼠 | Ift88 |
转基因 (插入) |
∙胆管形态异常 ∙肝纤维化 |
∙肾囊肿 ∙肾脏肿大 |
∙胰腺囊肿 ∙多指畸形 |
[ 87,88 ] |
| 韓國 | 老鼠 | Bicc1 |
化学 诱导 |
∙胆管扩张 |
∙肾囊肿 ∙肾小管扩张 |
∙胰管扩张 | [ 85 ] |
| ARPKD 模型 | |||||||
| 蛋白激酶C | 鼠 | PKHD1 |
自发 (剪接 突变) |
∙肝囊肿 ∙胆管扩张 ∙肝纤维化 |
∙肾囊肿 ∙肾脏肿大 ∙肾纤维化 |
∙胰腺囊肿 | [ 89,98 ] |
| Pkhd1ex40 | 老鼠 | PKHD1 |
有针对性 (插入击倒) |
∙肝囊肿 ∙肝纤维化 |
∙无 | ∙门脉高压症 | [ 90 ] |
| Pkhd1 del2/del2 ( Pkhd1 tm1Cjwa ) * | 老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙肝囊肿 ∙胆管扩张 ∙肝纤维化 |
∙肾囊肿 ∙肾小管扩张 |
∙胰腺囊肿 ∙胰管异常 |
[ 99 ] |
| Pkhd1 del3−4 ( Pkhd1 tm1 . 1Ggg ) * | 老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙肝囊肿 ∙肝纤维化 |
∙肾囊肿 ∙肾纤维化 |
∙胰腺囊肿 ∙胆总管囊肿 ∙升行性胆管炎 |
[ 60 ] |
| Pkhd1 del4/del4 ( Pkhd1 tm1Som )* | 老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙肝囊肿 ∙胆管囊肿 ∙肝纤维化 |
∙无 |
∙胰腺囊肿 ∙胰腺纤维化 ∙脾脏肿大 |
[ 100 ] |
|
Pkhd1 e15GFP ∆ 16 ( Pkhd1 tm1Gwu ) * |
老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙肝囊肿 ∙肝纤维化 |
∙肾囊肿 ∙肾小管扩张 ∙肾纤维化 |
∙胰腺囊肿 ∙胰管扩张 ∙胃肠道溃疡 |
[ 101 ] |
|
Pkhd1 lacZ (Pkhd1 tm1Sswi)* |
老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙胆管扩张 ∙肝纤维化 |
∙肾囊肿 ∙肾小管扩张 ∙肾纤维化 |
∙胰腺囊肿 | [ 102 ] |
| Pkhd1 LSL(-) ( Pkhd1 tm2Cjwa ) * | 老鼠 | PKHD1 |
有针对性 (插入 击倒) |
∙肝囊肿 ∙肝纤维化 |
∙肾囊肿 ∙肾小管扩张 |
∙未知 | [ 103 ] |
| Pkhd1 Δ67 | 老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙无 | ∙无 | ∙无 | [ 67 ] |
|
Dzip1l wpy/wpy ( Dzip1l warpy ) * |
老鼠 | Dzip1l |
靶向 (单点突变导致 KO) |
∙胆管形态异常 |
∙肾囊肿 ∙肾小管扩张 |
∙多指畸形 ∙眼部形态异常 ∙上唇裂 ∙腭裂 |
[ 37 ] |
|
Pkhd1 C642* ( Pkhd1 em1Mrug ) * |
老鼠 | PKHD1 |
被针对 (通过删除 KO) |
∙肝囊肿 ∙胆管囊肿 ∙肝纤维化 |
∙扩张的肾小管 ∙近端 小管扩张 |
∙未知 | [ 67 ] |
在单独的窗口中打开
* 模型名称根据 MGI (小鼠基因组信息学) [ 104 ] 命名。Ref. = 参考。KO = 敲除。
21 世纪初,随着PKHD1被发现为 ARPKD 的主要基因 [ 47 , 48 ],第一种 ARPKD 动物模型出现。多囊肾大鼠或 PCK 大鼠最初被提议作为 ADPKD 模型,因为其肾脏和肝脏疾病进展缓慢 [ 89 ],但后来证实PCK 大鼠的Pkhd1基因被破坏 [ 48 ]。第一个基于Pkhd1的转基因小鼠模型是Pkhd1 ex40 [ 90 ],后来出现了许多其他模型,以及基于Dzip1l的模型(表格1)。值得注意的是,肝脏 ARPKD 样表型在所有基于Pkhd1的模型中始终存在,但肾脏表型通常不存在。有趣的是,与 ARPKD 患者不同,这些模型中经常出现胰腺囊肿 [ 6 ]。
ARPKD 中囊肿形成的关键或主要分子机制仍不清楚。尽管如此,动物模型让我们能够扩展对疾病不同阶段的理解,从囊肿形成到囊肿进展。在这个复杂的过程中,已经描述了几种改变的分子通路,例如液体分泌、细胞异常增殖(如 mTOR、RAS-RAF-ERK 和 AKT)、由 PKA 激酶和 AC6 调节的 cAMP 通路、细胞外基质 (ECM) 的改变等 [ 91,92 ]。因此,近几十年来,由于存在多种动物模型,对 ARPKD 病理生理学的理解有所提高。然而,ARPKD 中囊肿形成的关键内在分子机制仍然未知,这导致人们对了解疾病机制和开发新治疗策略的兴趣日益浓厚。接下来,我们将回顾一下 ARPKD 中的主要分子通路。
7.2. EGFR轴表达和液体分泌异常
1992 年,首次有证据证明 PKD 中表皮生长因子受体 (EGFR) 轴发生了改变,当时证明 PKD 患者原代培养细胞可增加囊肿扩张 [ 105 ]。随后,在从 ADPKD 患者分离的原代细胞中,表皮生长因子 (EGF) 刺激囊肿形成 [ 106 ]。对于 ARPKD,首批数据来自cpk小鼠模型肾脏提取物,结果显示 EGF 表达上调 [ 107 ]。逐渐地,其他证据显示 EGFR 在体外 [ 108 ]、小鼠模型 [ 88,109,110 ] 和ARPKD患者 [ 111,112 ]中发挥重要作用,其中 EGFR 上调位于囊性上皮表面。同样,已证实 ARPKD 中存在 EGF[ 113,114] 和转化生长因子-α (TGFα)[115 ]的异常表达,且 EGFR 受体家族的几个成员 (EGFR1、ErbB2 和ErbB4 ) 在 ARPKD 啮齿动物模型中被发现过度表达[ 72,116 ] (图 3A)。这种过表达包括 mRNA、蛋白质和受体活性或磷酸化的增加[ 92 ]。此外,动物模型证据表明肝囊肿形成中 EGFR 轴存在类似的异常[ 117 ]。
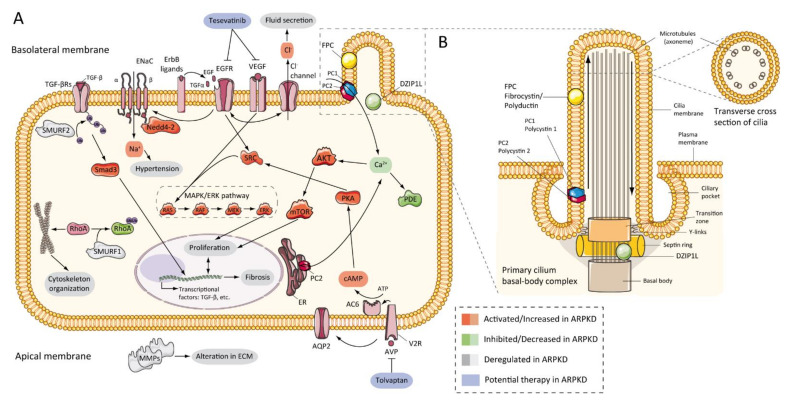
图 3
ARPKD 分子发病机制:( A ) 图表代表 ARPKD 肾上皮细胞中可能上调、下调或失调的通路以及可能的潜在疗法;( B ) 卡通表示 ARPKD 蛋白在肾上皮细胞纤毛中的定位。FPC 位于纤毛的初级顶端和基底体区域,而 DZIP1L 位于纤毛基底体的过渡区。 (FPC—纤维囊蛋白/多聚导管蛋白;DZIP1L—DAZ 相互作用锌指蛋白 1 样;PC1—多囊蛋白 1;PC2—多囊蛋白 2;TGF—转化生长因子;ENaC—上皮钠通道;Na + —钠阳离子;EGFR—表皮生长因子受体;VEGF—血管内皮生长因子;Cl − —氯阴离子;SMURF—SMAD 特异性 E3 泛素蛋白连接酶;Nedd4-2—E3 泛素蛋白连接酶 Nedd4-2;SRC—原癌基因酪氨酸蛋白激酶 Src;AKT—RAC-alpha 丝氨酸/苏氨酸蛋白激酶;Ca + —钙阳离子;PDE—磷酸二酯酶;mTOR—雷帕霉素的哺乳动物靶点;MAPK—丝裂原活化蛋白激酶;RAF—快速加速纤维肉瘤;MEK—丝裂原活化蛋白激酶激酶;ERK—细胞外信号调节激酶;Rhoa—Ras 同源物家族成员 A;ER—内质网;PKA—蛋白激酶 A;cAMP—环磷酸腺苷(环 AMP);ATP—三磷酸腺苷;AC6—腺苷酸环化酶 6 型;AQP2—水通道蛋白 2;V2R—加压素受体 2;AVP—精氨酸加压素;MMPs—基质金属蛋白酶)。
EGFR信号传导与囊肿形成有关,EGFR酪氨酸激酶活性上调与囊肿生长之间存在相关性。通过点突变降低EGF酪氨酸激酶活性的改良orpk模型(wa2小鼠)[ 118 ]显示囊肿形成显著减少、肾功能改善[ 110 ] 。此外,使用EGFR特异性酪氨酸激酶抑制剂进行药物抑制可降低EGFR活性,从而显著减缓囊肿进展[ 119、120、121 ]。有争议的是,EGFR酪氨酸激酶抑制剂并没有减缓PCK大鼠肾囊肿的进展[ 122 ]。
上皮分泌是囊肿形成的关键病理生理学组成部分。ARPKD 净液体分泌中钠摄取减少被认为与上皮钠通道 α 亚基中 EGF 减少有关 [ 123 , 124 ]。然而,关于这一主题已经发表了相互矛盾的数据 [ 116 ]。在来自 ARPKD 患者囊肿的细胞中,有人认为钠的吸收是由上皮钠通道 (ENaC) 介导的 [ 125 ] 。此外,该机制被认为是 ARPKD 中高血压的调节剂 [ 125、126、127 ]。
另一方面,在 ADPKD 中,数据显示 Cl −分泌通过囊性纤维化跨膜传导调节器 (CFTR) 发生 [ 128 , 129 ]。然而,在 ARPKD 中,这种机制没有明显的相关性。在bpk小鼠模型中,CFTR 的缺失并未减缓肾囊肿和肝囊肿的进展 [ 130 ]。这些数据表明,ADPKD 和 ARPKD 中 Cl −和液体分泌的机制不同。
7.3. cAMP 与增殖
多项研究表明,腺苷酸环化酶腺苷 3′,5′-环磷酸腺苷 (cAMP) 通路可刺激 ARPKD 和 ADPKD 肾上皮细胞增殖。囊肿上皮中 cAMP 生成异常,导致囊液中含有大量该核苷酸 [ 122 , 131 , 132 , 133 ]。cAMP 可激活 ADPKD 肾囊肿上皮 [ 134 , 135 , 136 ] 以及培养的 ADPKD 和 ARPKD 细胞 [ 133 ] 中的 B-Raf、MEK 和 ERK 通路。同样,这些结果还得到了一些数据的补充,这些数据显示,在几种 ARPKD 啮齿动物模型中,MAPK 和 AKT/mTOR 通路在蛋白质水平上呈上调趋势 [ 137、138、139、140、141 ]。这些事实与细胞内 Ca 2+的减少和 SCR 蛋白的磷酸化有关 [ 142、143、144 ]。尤其是,阻断细胞内 Ca 2+可提高培养的 ARPKD 细胞中的 AKT 和增殖活性 [ 143 ]。这项研究为将细胞内钙水平恢复作为 PKD 的治疗方法提供了机会。
PKD 中钙离子失调会导致血管加压素 V2 受体上调 [ 136 , 138 ],从而激活 cAMP/PKA 级联 [ 145 , 146 ]。在一项临床前试验中,V2 受体拮抗剂已证明其在 ARPKD 大鼠模型中有效,可降低肾脏 cAMP 水平并改善囊性肾病 [ 137 ](图 3A)。这一事实已在临床前 [ 147 , 148 ] 和临床水平 [ 149 ] 上得到进一步研究、审查和理解,以至于托伐普坦(一种加压素 V2 受体抑制剂)已获准用于人类患者 [ 150 ],并且正在进行针对 ADPKD 儿童的试验(托伐普坦 ( NCT02964273 ))[ 151 ]。ARPKD 相关的临床试验将在后面进行审查。
7.4. ARPKD 病理生理学中涉及的其他途径
在 ARPKD 中还发现了其他致病特征,以及细胞外基质 (ECM) 和金属蛋白酶 (MMP) 表达的改变[ 152 , 153 ]、 Pkhd1缺陷细胞中血管内皮生长因子 (VEGF) 和缺氧诱导因子-1α (HIF-1α) 的上调[ 139 ]、动物模型中过氧化物酶体增殖激活受体-γ (PPAR-γ) 的上调[ 154 , 155 ]或代谢改变[ 156 ]。在一项大型而有趣的研究中,Kaimori 和他的同事发表了有关 FPC 与 C2-WWW-HECT 结构域 E3 泛素连接酶家族成员之间的新功能关系的信息。作者将 FPC 定位在 Ndfip2 也存在的囊泡中,Ndfip2 是一种泛素连接酶相互作用蛋白,参与运输和调节 Nedd4-2 泛素连接酶家族以及 SMURF1 和 SMURF2。这些数据可能通过 TGF-β 信号通路解释 ARPKD 和肾脏和肝脏纤维化中的不同通用表型,通过 ENaC 介导的钠重吸收解释高血压,通过 RhoA 泛素化和细胞骨架组织解释囊肿形成 [ 127 ](图 3A). 其他研究认为,PKD 中的管状形态发生与平面细胞极性 (PCP) 异常有关 [ 157 ]。然而,后来的研究却得出了相反的结果 [ 158 ]。
7.5 纤毛的作用
图 3图 2 显示 ARPKD 蛋白(锌指蛋白 DZIP1L 和 FPC)位于纤毛中。纤毛是从哺乳动物细胞表面发出的长而微管的结构。初级纤毛的轴丝含有 9 个外周微管束(9 + 0 型)。与正常纤毛功能丧失相关的病理称为纤毛病,包括 ARPKD [ 159 ]。PC2 也称为 TRPP2,是瞬时受体通道 (TRP) 家族的成员,是一种钙通透性非选择性通道 [ 160 ]。PC2和 PC1 形成受体-通道复合物,参与钙通路和纤毛反应 [ 161、162、163 ](图 3B)。研究表明,FPC 能与初级纤毛中的 PC2 相互作用并调节 PC2 通道活性[ 101、164、165 ]。此外,有报道称,FPC 的 C 端在体内和体外与 PC2 的 N 端发生物理相互作用,而Pkhd1缺陷细胞则表现出 PC2 通道活性失调[ 101 ]。然而,Wang 等人发现,在 FPC 水平降低的细胞中,PC2 水平没有差异[ 165 ]。使用新型Pkhd1小鼠模型的其他数据显示,删除Pkhd1的最后一个外显子、PC2 结合位点和核定位信号对小鼠没有明显的病理影响[ 67 ]。此外,研究人员无法在转基因小鼠模型的肾脏样本中共沉淀 FPC-PC2。这些结果表明,FPC 的 PC2 结合结构域对于纤维囊蛋白的功能并非必需的 [ 67 , 166 ]。
我们已描述了Pkd1和Pkhd1之间的遗传相互作用,将 ADPKD 和 ARPKD 联系在一起 [ 60 ]。因此,其他数据报告了其他啮齿动物模型中Pkd1和Pkhd1之间的这种遗传相互作用,描述了Pkd1和/或Pkhd1的轻度囊性疾病表型结合后其严重程度增强 [ 38 , 167 ]。这些研究和其他研究扩展了 FPC 和 PC1 之间的关系。使用体外模型,FPC 的缺失不会影响 PC1 的生物合成和定位,这表明Pkd1和Pkhd1之间的遗传相互作用是间接的 [ 30 , 167 ]。
这些研究结果似乎与以下观点一致:PKD 蛋白在纤毛中形成具有共同下游信号通路的功能性复合物 [ 168 ]。有趣的是,纤毛丢失会抑制 ADPKD 小鼠模型和常染色体显性多囊肝病 (ADPLD) 小鼠模型中的肾囊肿生长 [ 169 ]。然而,在最近的一项研究中,Gallagher 和 Somlo 报告称,纤毛的丢失并不能减缓 ARPKD 中肝病的进展 [ 170 ]。这些数据表明,至少在肝囊性表型中,ADPKD 和 ARPKD 没有共同的纤毛相关通路。
另一方面,根据多项细胞培养研究、斑马鱼和小鼠的研究,DAZ 相互作用蛋白 1 样蛋白 (DZIP1L) 定位于中心粒和纤毛过渡区初级纤毛 [ 37 ]。Lu 等人证明了 DZIP1L 与 septin 2 (SEPT2) 之间的相互作用,SEPT2 是一种参与维持过渡区纤毛周围扩散屏障的蛋白质 [ 171 ],并且与纤毛过渡区蛋白 tectonic 1 (TCTN1) 共定位。在DZIP1L突变细胞中,多囊蛋白-1 和 -2 从基体到纤毛轴丝的运输发生改变;当它们正常分布在纤毛轴丝中时,两者都保留在基体中。然而,DZIP1L缺乏不会改变 FPC 的定位或表达 [ 37 ]。这些发现表明DZIP1L在多囊蛋白的运输中发挥着作用,并提供了将 ARPKD 与 ADPKD 联系起来的新证据。
去:
8.临床试验
正如我们所指出的,PKD 中囊肿形成的核心或关键机制仍不清楚。细胞和动物研究表明,PKD 的发病机制涉及多种途径。这些事实使得多种药物进入临床阶段(表 2)。
表 2
目前正在进行或已完成 ARPKD 临床试验。
| 标识符 | 干涉 |
研究设计 和特征 |
研究描述 | 赞助 |
|---|---|---|---|---|
| NCT04782258 | 托伐普坦 |
∙研究类型:干预 ∙主要目的:治疗 ∙时间:2021 年 4 月 - 2025 年 6 月(预计) ∙患者:20 人(预计,尚未招募) ∙分配:非随机 ∙干预模型:平行分配 ∙掩蔽:无(开放标签) |
∙本次 3 期试验的主要目的是评估托伐普坦 (OPC-41061) 对 8 天至 18 岁以下 ARPKD 婴儿和儿童的安全性。 ∙本次研究的参与者将被分配使用托伐普坦 18 个月,并在研究过程中受到密切监测。 |
大冢 制药开发与商业化有限公司, 美国新泽西州普林斯顿。 |
| NCT04786574 | 托伐普坦 |
∙研究类型:干预 ∙主要目的:治疗 ∙时间:2021 年 4 月 - 2025 年 7 月(预计) ∙患者:20 人(预计,尚未招募) ∙分配:N/A ∙干预模式:单组分配 ∙掩蔽:无(开放标签) |
∙在本次 3 期试验中,主要目标是评估托伐普坦 (OPC-41061) 对 28 天至 12 周以下 ARPKD 儿科受试者的安全性、耐受性和有效性。 ∙本次试验的参与者将被分配使用托伐普坦 24 个月,并在研究过程中受到密切监测。 |
大冢 制药开发与商业化有限公司, 美国新泽西州普林斯顿。 |
| NCT03096080 | 替塞伐替尼 |
∙研究类型:干预 ∙主要目的:治疗 ∙时间:2017 年 3 月 - 2019 年 10 月(已完成) ∙患者:10 ∙分配:非随机 ∙干预模型:顺序分配 ∙掩蔽:无(开放标签) |
∙第 1 阶段的这项试验评估了 Tesevatinib (KD019, XL647) 液体制剂单次递增剂量给药于 ARPKD 儿科受试者(5-12 岁儿童)的安全性和耐受性。 ∙为了确定 Tesevatinib 液体制剂对 ARPKD 儿科受试者的安全性,所有参与者在研究入组第一天均接受了活性研究药物。 |
Kadmon Corporation, LLC 美国宾夕法尼亚州费城。 美国威斯康星州密尔沃基。 |
| NCT01401998 | 观察 |
∙研究类型:观察性 ∙时间:2011 年 7 月 - 2022 年 12 月(招募) ∙患者:200 人(预计) ∙观察模型:队列 ∙时间视角:回顾性任务 |
∙本研究收集了 ARPKD 患者的临床和遗传信息,以扩大对疾病的了解。 ∙本试验的主要目标是创建一个临床和突变数据库,其中包括临床信息和识别所有参与研究的患者的基因突变。 ∙突变数据库将有助于促进基因研究,如基因型-表型相关性、新疾病基因研究或修饰基因研究。 ∙使用受影响个体和对照个体的人体组织创建组织资源。 |
Lisa M. Guay-Woodford (合作者: 国家糖尿病、消化和肾脏疾病研究所 (NIDDK))。 美国哥伦比亚特区华盛顿。 |
| NCT00068224 | 观察 |
∙研究类型:观察性 ∙时间:2003 年 9 月 - 2021 年 2 月(已完成) ∙患者:374 ∙观察模型:队列 ∙时间视角:前瞻性 |
∙本研究评估了患有纤毛病(包括 ARPKD)的患者。 ∙本研究的目的是更好地了解这些疾病的医学并发症,并找出有助于设计新疗法的特征。 |
美国国家人类基因组研究所 (NHGRI)。 美国马里兰州贝塞斯达。 |
在单独的窗口中打开
状态根据https://clinicaltrials.gov/,于 2021 年 4 月 6 日访问。
根据 3 期和 4 期临床试验的结果 [ 149 , 172 , 173 ],目前正在进行两项使用托伐普坦药物干预的临床试验。这些试验的主要目的是评估托伐普坦对婴儿(8 天或以下)和儿童(18 岁以下)(NCT04782258)以及儿科患者(28 天至 12 周龄)(NCT04786574)的安全性。另一方面,PKD 表现出异常的 c-Src 活性,连接 cAMP 和 EGFR 分子途径 [ 174 ],并且其抑制可改善肾囊肿形成 [ 144 ]。这些数据促使 Sweeney 等人。研究 EGFR 轴、c-Src 和 VEGFR 多激酶抑制剂特塞瓦替尼 (TSV) 作为 ARPKD 临床前研究中可能治疗方法的效果,并获得了良好结果 [ 175 ]。积极而有希望的结果促使 ARPKD TSV 临床试验 I 期和 II 期获得批准 ( NCT03096080 )。最后,正在进行两项观察性试验,以扩大对该疾病的了解(基因型-表型相关性、临床方面)并创建更精确的突变和临床数据库(NCT01401998和NCT00068224)。
去:
9. 结论
ARPKD 领域在遗传学、诊断学和分子生物学领域取得了重大进展。首先,鉴定出多年来被认为是唯一 ARPKD 基因的PKHD1基因,最近又鉴定出DZIP1L基因,并将其应用于基于下一代测序 (NGS) 的基因诊断。其次,对纤维囊蛋白/多聚胆管蛋白的功能和定位的理解取得了进展。最后,不同的研究(尤其是在动物模型中)开始阐明与该疾病的发病机制有关的途径,并确定可能的治疗方法。
然而,许多未解问题需要在未来得到解答。新发现的DZIP1L作为 ARPKD 的第二个基因位点,为出现导致 ARPKD 的新基因留下了可能性,在这方面,有必要应用基因未解析的全外显子组测序 (WES) 家族 (GUR) 来研究 ARPKD 表型。FPC 的确切功能仍然未知,其所有亚型的正确表征也尚不清楚。重要的是,驱动 PKD 囊肿形成的关键因素尚不清楚。由于这些原因,ARPKD 的致病性尚不清楚,即使在今天,现有的替代疗法也没有获批的治疗方法。为了解释 ARPKD 的遗传和细胞基础,对该主题的研究成为摆脱这种困境的唯一出路。
(责任编辑:佳学基因)
