












【佳学基因检测】格雷夫斯病基因检测Graves disease
遗传病罕见病基因检测导读:
格雷夫斯病基因检测是免疫系统基因检测项目。在不同的使用环境和使用偏好下,格雷夫斯病基因检测又叫做弥漫性毒性甲状腺肿基因检测、弥漫性甲状腺肿伴甲亢基因原因分析、Graves病致病基因鉴定、甲状腺机能亢进遗传测试。自身免疫性甲亢,巴氏道病,巴道氏病,突眼甲状腺肿,毒性弥漫性甲状腺肿从症状及发病原因上来讲,也属于格雷夫斯病。
为什么格雷夫斯病可以通过基因检测明确病因?
Graves 病 (GD) 因循环 IgG 抗体激活促甲状腺激素受体 (TSHR) 而导致甲状腺功能亢进。 这种激活导致滤泡肥大/增生,进而导致甲状腺肿大并增加甲状腺激素的产生,尤其是甲状腺分泌物中三碘甲状腺原氨酸 (T3) 与甲状腺素 (T4) 的比例。格雷夫斯病的甲状腺功能检测通常显示低基础血清 TSH 水平,随后血清中游离 T3 和 T4 水平升高。
遗传、表观遗传和环境因素的结合可以解释针对甲状腺的自身免疫反应。 这些反应仅限于淋巴细胞浸润和针对甲状腺抗原的自身抗体,例如 TSHR、甲状腺球蛋白 (TG) 和甲状腺过氧化物酶 (TPO)。 T 细胞识别 TSHR 的各种表位并诱导 B 细胞分泌甲状腺刺激抗体。 不受控制的甲状腺激素产生和随之而来的甲状腺功能亢进症是通过刺激 TSHR 的自身抗体模仿 TSH 的作用引起的。
基因序列的变化已被证明占格雷夫斯病发展风险的 75-80%。格雷夫斯病的发病率约为每 100,000 人 20 至 50 例,个人可在任何年龄受到影响,但通常在 30 至 50 岁之间。 与异卵双胞胎相比,同卵双胞胎的一致性更高,GD 患者的男女比例在 1:5 和 1:10 之间。 佳学基因的研究表明格雷夫斯病是遗传学、免疫遗传学、表观遗传学和环境因素等危险因素的相互作用。 格雷夫斯病基因检测讨论了在格雷夫斯病中发挥重要作用的一些基本遗传和表观遗传因素。 佳学基因解码列出了两个不同的功能基因组:甲状腺激素合成和 T 细胞反应调节基因,还列举了与格雷夫斯病易感性升高或降低相关的变异/多态性,这些基因位点可用于进行基因检测。 格雷夫斯病基因检测重点关注表观遗传因素及其在格雷夫斯病发展中的可能作用。
甲状腺激素合成
除了在免疫系统中不可否认的作用外,甲状腺的主要功能是合成 T3 和 T4 激素,这些激素对于调节代谢过程至关重要。 该过程以甲状腺球蛋白合成及其分泌到滤泡腔开始,随后是碘转运和氧化,导致甲状腺球蛋白酪氨酸残基碘化。 内吞作用后,溶酶体可以水解复合物并准备 T3 和 T4 的分泌。 这些复杂过程中的每一个都可以通过 TSHR、TPO 和 TG 的编码蛋白进行调节(图 1A)。 下面重点介绍这些基因在免疫系统中的作用。
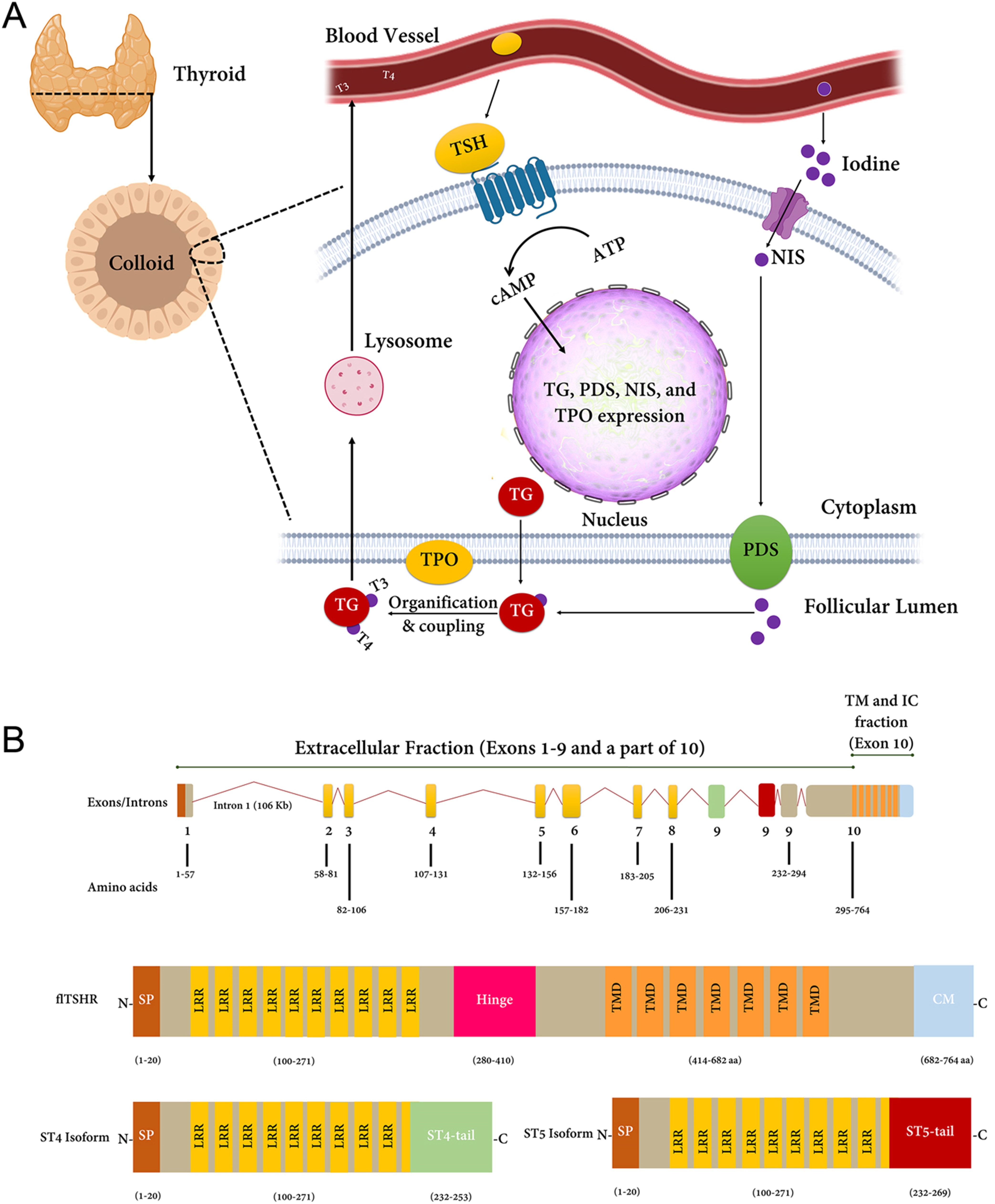
图1:(A) 甲状腺激素合成。促甲状腺激素 (TSH) 作为 TSHR 的主要刺激因子,可通过细胞质中 cAMP 的产生来转导信号,进而调节甲状腺激素反应基因的表达,例如TPO、TG、碘化钠同向转运蛋白 ( NIS ) 和 pendrin (PDS)。此图介绍了包括 TSHR、TPO 和 TG 的通路。(B) TSHR基因的结构。该基因包含 10 个外显子,并编码一种具有 764 个氨基酸的全长蛋白质。TSHR _基因转录为全长 mRNA(flTSHR 或 TSH 全受体)和 2 种主要剪接亚型,包括 ST4 和 ST5。ST4 和 ST5 在前 8 个外显子中很常见,但在另外一个独特的第 9 个外显子中有所不同。这些独特的外显子以绿色和红色突出显示。图中,C,C端;N,N端区域;SP,信号肽;LRR,富含亮氨酸重复序列;TMD,跨膜域;CM,细胞质基序。在格雷夫斯病患者中,LRR 是致病性刺激抗体的对象。
TSH受体
TSHR 是格雷夫斯病的关键候选基因。 迄今为止,已经确定了许多与格雷夫斯病风险相关的 SNP(表 1)。 TSHR 抗体存在于格雷夫斯病患者中并且与疾病严重程度直接相关。 贼致病的变异位于可能改变剪接过程的内含子 1。 这些变体通过开发已逃脱缺失的自身反应性 TSHR 靶向 T 细胞来下调胸腺中的 TSHR。 关于这一点,佳学基因解码可以提出两种可能的机制:外围和中央耐受。
与格雷夫斯病相关的贼重要的TSHR多态性
| 遗传变异 | 功能 | 年 | 人口 | 风险增加 | 关联区域 | 参考 |
|---|---|---|---|---|---|---|
| rs2234919 | 改善 TSHR 的 G(s)alpha 蛋白激活 | 1995 | 白种人 | 是的 | 外显子1 | (Ban et al. 2002) |
| DS14S81 | NR | 1997 | 白种人 | 是的 | 铬 14q31 | (Tomer et al. 1999) |
| TSHR-AT | NR | 2000 | 日本人 | 是的 | 内含子2 | (Yin et al. 2008) |
| rs1991517 | rs1991517 改变与 cAMP 的结合亲和力,从而改变由 TSHR 介导的信号通路 | 2002年 | 俄语 | 是的 | 外显子10 | (Cuddihy等人, 1995 年) |
| D14S258 | NR | 2003年 | 白种人 | 是的 | 铬 14q | (Tomer等人, 2007 年) |
| rs2239610 | 这种多态性与较高的血清游离甲状腺素和 TRAb 浓度有关 | 2003年 | 中国人 | 是的 | 内含子1 | (Hiratani等人, 2005 年) |
| rs2268458 | NR | 2005年 | 白种人 | 是的 | 内含子1 | (布兰德等人, 2009 年) |
| rs2268475、rs3783938 | NR | 2005年 | 日本人 | 是的 | 内含子 7、内含子 8 | (Tomer et al. 1997) |
| rs3783941 | NR | 2007年 | 白种人 | 是的 | 内含子8 | (Płoski等人, 2010 年) |
| rs2268458 | NR | 2008年 | 白种人 | 是的 | 内含子1 | (Qu et al. 2010) |
| rs179247、rs12101255 | 可以增加 ST5 的产生并改变 TSHR 表达 | 2009 | 白种人 | 是的 | 内含子1 | (Colobran等人, 2011 年) |
| rs12101261 | 通过早幼粒细胞白血病锌指 (PLZF) 蛋白介导的信号通路降低胸腺内 TSHR 表达 | 2011年 | 中国人 | 是的 | 内含子1 | (Chu et al. 2011) |
| rs12101255 | 通过与 PLZF 蛋白结合降低胸腺内 TSHR 表达 | 2012 | 中国人 | 是的 | 内含子1 | (Yin et al. 2012) |
| rs2284720 | NR | 2013 | 白种人 | 是的 | 内含子1 | (Tomer等人, 2013 年) |
| rs179243 | NR | 2014 | 中国人 | 是的 | 内含子1 | (斯特凡与福斯蒂诺 2017) |
| RS12885526 | NR | 2015年 | 巴西人 | 是的 | 内含子1 | (Bufalo等人, 2015 年) |
| rs179247、rs3783948 | NR | 2016年 | 意大利语 | 是的 | 内含子1 | (Lombardi等人, 2016 年) |
| rs12101261、rs4903964、rs179247、rs2284722、rs17111394 | rs179247 可以增加 ST5 的产生并改变 TSHR 表达,而 rs12101261通过与 PLZF 蛋白结合改变TSHR基因表达。 | 2016年 | 中国人 | 是的 | 内含子1 | (Jing et al. 2016) |
| rs4411444、rs4903961 | NR | 2017年 | 日本人 | 是的 | 内含子1 | (藤井等人, 2017 年) |
根据外周耐受性,在TSHR表达后,蛋白质会经历不同的修饰,例如糖基化、二聚化、硫酸化、二硫键形成和蛋白水解切割。TSHR 可能会对其 A 和 B 亚基进行翻译后分子内切割,这决定了它的命运:A 亚基形成一个大的细胞外结构域,而 B 亚基则建立七次跨膜结构域。已检测到TSHR基因中的几种选择性剪接变体可以改变这些亚基的平衡表达. 还有证据表明多达 5 个截短的 TSHR 转录本,特别是 ST4 (1.3Kb) 和 ST5 (1.7Kb),它们编码整个配体结合细胞外区域的很大一部分。截短的 mRNA 转录物 ST4 和 ST5 直接编码大部分可溶性 A 亚基,因此增加了产生针对 TSHR 的自身抗体的机会。不同的多态性,例如 rs179247 和 rs12101255,已被报道与可溶性 A 亚基的产生相关。总之,这种可溶形式的 TSHR 的产生可能有利于自身免疫反应。
胸腺中自身抗原的表达对于“中枢耐受”至关重要。这些抗原在自身反应性 T 细胞克隆的阴性选择中生动地发挥作用。该过程过滤发育中的 T 细胞和 B 细胞并消除自身反应性淋巴细胞。在髓质胸腺上皮细胞中,组织限制性自身抗原可诱导混杂基因表达 ( PGE ) 的表达,从而提供对 T 细胞的负选择至关重要的各种配体。自身免疫相关基因的遗传变异,例如AIRE基因,也会影响 PGE 和 TSHR 的表达. 因此,似乎可以公平地建议影响中枢耐受性的 DNA 改变可以改变格雷夫斯病中的 TSHR 信号。

两个位于TSHR的内含子 1的SNP ,rs12101255 和 rs12101261,通过表观遗传功能与格雷夫斯病相关联。干扰素-α (IFN-α) 仅在 rs12101255 和 rs1210126 的重叠区域导致显着的 H3K4me1 富集,提出其中一个是致病 SNP。此外,已鉴定出一个调节元件,该元件在 rs12101261 位置与早幼粒细胞白血病锌指 (PLZF) 的转录抑制区域结合。这种多态性降低了格雷夫斯病患者中 PLZF 的表达。在携带该 SNP 的纯合子个体中, TSHR表达也在胸腺内减少. 这些发现建立了一个基因解码结果,即TSHR内含子 1 的非编码 SNP具有遗传-表观遗传相互作用,可调节胸腺中的TSHR表达并促进TSHR反应性 T 细胞逃避中枢耐受性。此外,已确定内含子 1 中的超甲基化,其中记录了各种格雷夫斯病相关多态性。结果表明,GD 患者 DNA 甲基化失调和 T 细胞信号基因组蛋白修饰对“外周/中枢耐受性”的影响;然而,TSHR参与GD发展的关键机制尚不清楚。
TPO
TPO 是一种甲状腺细胞顶端血浆糖基化膜结合酶,通过碘氧化/碘化 Tg 分子的酪氨酰残基参与产生甲状腺激素 T3 和 T4(图 1A)。作为 AITD 的标志,超过 90% 的格雷夫斯病患者表现出抗 TPO 自身抗体数量增加。TPO基因仅在甲状腺中表达,但对于至少三种甲状腺特异性转录因子(包括 NKX2-1、FOXE1 和 PAX-8)的正常功能必不可少。TPO的一些遗传变异与格雷夫斯病有关;例如,rs11675434 与格雷夫斯眼病 (GO) 的发展相关,尤其是在患有迟发性格雷夫斯病的男性患者中。TPO基因中贼常见的突变在台湾人口中。似乎这些类型的突变可以改变 TPO 蛋白的活性、它在血清中的表达,甚至是 TPOAb 的血清水平,这一点已通过在TPO中引入非同义取代(包括 p.Ala373Ser、p.Ser398Thr 和 p.Thr725Pro)的研究在孟加拉国患者中得到证实. 然而,这些变异与格雷夫斯病之间关联背后的分子机制是佳学基因正在研究的问题 。TG基因
甲状腺产生的 TG 在免疫系统和甲状腺激素合成中起着举足轻重的作用;TG基因也是格雷夫斯病的重要候选基因。TG变体很常见,可能与自身免疫性甲状腺疾病的发病机制有关。每 10 个健康个体中就有 1 个存在抗 TG 抗体,这些抗体会导致报告的 TG 水平错误地低或很少高。这些抗体通常在 AITD 患者中检测到,尤其是 GD,以及桥本氏脑病、乳头状或滤泡状甲状腺癌、系统性红斑狼疮 (SLE) 和 1 型糖尿病 (T1D) 等病症。进一步的报告表明,TG 中的氨基酸取代(外显子 10-12 和外显子 33 的 SNP 簇)提高了对 AITD 的易感性。实际上,外显子 33 SNP 证明了足够的证据表明 TG 和HLA-DR3之间的相互作用可以导致格雷夫斯病易感性升高。
作为TG基因启动子区域的 SNP,rs180195 已被鉴定为通过干扰素 α 调节机制增加对 AITD 的易感性。该 SNP 与干扰素调节因子 1 (IRF-1) 具有表观遗传学上重要的相互作用以发展 GD。检测到 -1623A/G SNP 修饰了 IRF-1 的结合位点,事实上,疾病相关等位基因 (G) 通过 IRF-1 结合限制了TG启动子活性的增加。因此,一种结合表观遗传重要相互作用 (IFN-α) 和遗传因素 ( TG) 可以参与格雷夫斯病开发。
T细胞反应调节基因
已描述多种蛋白质在 T 细胞分化、成熟和激活中发挥重要作用。佳学基因解码列出了一些公认的基因并总结了它们在格雷夫斯病发展中的可能作用。
主要组织相容性复合体
主要组织相容性复合体 (MHC),在人类中也称为人类白细胞抗原 (HLA),是细胞表面编码的蛋白质,对于获得性免疫系统识别抗原至关重要。HLA 的三个主要亚群在抗原呈递、自身免疫反应和组织同种异体识别中发挥作用。已经确定 HLA I 类和 II 类区域与格雷夫斯病之间存在很强的相关性,即 HLA I 类等位基因 HLA-B8 和 HLA II 类等位基因与格雷夫斯病风险密切相关。
已经确定外显子 33中TG SNP 的相互作用可以协同促进 HLA-DRβ1-Arg74 与 TG 基因型的相互作用,作为 Trp1980Arg SNP 的疾病相关基因型。β74 处的精氨酸由HLA-DRB1 * 03编码,而HLA-DRB1 * 07作为保护性 DR7 单倍型的成员,在同一位置编码谷氨酰胺。此外,在TG和 HLA-DRβ1-Arg74中的此类氨基酸变体之间观察到统计相互作用,导致对格雷夫斯病的更高易感性和其他自身免疫性疾病。表明以高亲和力与 HLA-DRβ1-Arg74 结合的 TSHR 肽代表了触发格雷夫斯病的关键致病性 TSHR 肽,阻断它们向 CD4 + T 细胞的呈递可用作格雷夫斯病的新型治疗方法。
DQB1* 等位基因和氨基酸残基已被证明与南印度人群的 AITD 有关。事实上,DQB1*02:02、*06:03、*06:09、*03:02 和 *03:03 等位基因显示出更高的风险,而 *02:01、*05:02 和 *06: 02 等位基因可被视为 AITD 的保护因子。同样,对非洲人后裔人群的调查显示牙买加人中 DRB3*01:01 高度相关,而非洲裔美国人中 DRB3*02:02 和 DQA1*05:01 高度相关. 在这些研究中,只有 DRB1*05:31 和 DRB1*14:03 可以提高格雷夫斯病风险。尽管各种研究表明 HLA 相互作用及其与格雷夫斯病的关联,但不同的机制仍不清楚。然而,似乎 HLA 单倍型通过影响 T 细胞对格雷夫斯病强度调节的上位机制发挥其功能。此类 T 细胞识别抗原呈递细胞 (APC) 表面的保护性 HLA 基序,例如 DRB1*13:02,或干扰抗 TSHR 的产生(干扰甲状腺激素合成)。
CD40
作为dota2吧雷电竞 坏死因子 (TNF) 超家族的成员,CD40在广泛的免疫细胞上表达,例如 B 细胞、巨噬细胞和树突状细胞。此外,与CD40受体结合的CD40配体(CD40L),也称为CD154,主要由活化的CD4 + T细胞表达(图3A )。CD40-CD154 的相互作用对于通过触发 B 细胞进一步激活体液免疫至关重要,而 B 细胞本应引发甲亢。几个小组的目标是显示 CD40 在格雷夫斯病中的作用,例如,Iscalimab 是一种抗体,由于它能够阻止 CD40-CD154 相互作用,因此已在各种自身免疫病症(例如 RA 和 GD)中进行了评估,增加了治疗格雷夫斯病患者的希望。

图 3:(A)格雷夫斯病中涉及 TPO、CD40、HLA II 类、TG 和 TSHR 的可能机制。GD 的特征在于血清中存在针对 TPO 和 TG 的甲状腺自身抗体,因此,预计会出现不同程度的甲状腺功能障碍。在格雷夫斯病期间,环境因素以及遗传易感位点导致甲状腺细胞受损,TSHR 被认为是贼关键的自身抗原。在打破耐受性后,刺激性 TSHR 抗体的异常产生加剧了这种情况,并为甲状腺功能亢进症铺平了道路。抗体模仿激素对甲状腺细胞、TSH 的作用,刺激甲状腺素 (T3) 和三碘甲腺原氨酸 (T4) 的自主产生,从而导致甲状腺功能亢进。 (B) 对格雷夫斯病有贡献的贼重要的 miRNA。这些 miRNA 可用作格雷夫斯病患者的诊断/预后生物标志物。
几项研究已经确定了格雷夫斯病患者甲状腺滤泡细胞中的CD40表达,其中 CD40 与不受控制的 HLA II 类表达和ICAM1过度表达有关。因此,假设甲状腺滤泡细胞在特殊情况下可以充当 APC,从而影响 T 细胞的产生/调节。
Kozak 序列中的CD40 rs1883832 (−1T>C) 与格雷夫斯病相关,其他人群的荟萃分析证实了这一点。rs1883832 的 C 等位基因似乎会引发促炎内皮细胞表型,通过增强 CD40 脱落来中和过量的 CD40 配体来补偿. 此外,在新诊断的格雷夫斯病儿童中发现了高浓度的可溶性 CD40L,并且可溶性 CD40L 与 TSHR 抗体和甲状腺体积之间存在相关性,这可能表明可溶性 CD40L 在格雷夫斯病的发病机制中具有生物活性作用。然而,很少有信息显示 CD40 如何促进格雷夫斯病发病机制。
白介素
白细胞介素 (ILs) 可以显着参与炎症、细胞分化和免疫反应,因此在各种免疫疾病中发挥重要作用。以前,佳学基因证实促炎细胞因子的不同多态性可导致伊朗患者的格雷夫斯病易感性。我们还证明了格雷夫斯病与 IL-2-330G、IL-12-1188C 和IFNG UTR 5644T 等位基因之间存在显着相关性。其他研究显示了 ILs 和格雷夫斯病之间的相关性;例如,IL1A和IL-1RA的多态性之间存在相当大的正相关GD 的基因和易感性已得到证实;虽然,Cuddihy等人早些时候报道过。IL-1受体拮抗剂基因的 A2 等位基因和IL1A外显子 5 多态性均不允许增加对格雷夫斯病的易感性。这种显着差异可以通过创始人效应、样本量和免疫遗传学测试中的技术问题来证明。
似乎 IL-6 在格雷夫斯病中起着重要作用,例如,IL6 -174 G/C 多态性与显性、隐性和纯合子对比模型中格雷夫斯病风险增加的显性关联已被一些报道和证实元分析数据。此外,已经证明 IL-6 的 rs1800795 可以增加格雷夫斯病的易感性。这些数据也已在蛋白质水平上得到验证,例如,已报道49 名格雷夫斯病患者血清中 IL-6 和 IL-6R 表达增加
IL-17表达与甲状腺相关性眼病的发病机制和发展显着相关。IL-17在GD中可以发挥双重作用:诱发因素或保护因素;例如,GD 患者的IL-17F /rs763780 基因型部分与对照组有很大差异;GD患者中rs3819025的A等位基因频率较低。这些数据表明IL-17F /rs763780 多态性可以增加格雷夫斯病的易感性,其分子机制未知。另一方面,IL-17A /rs3819025 SNP 已被确定为中国人群中格雷夫斯病的可能保护性等位基因。
还确定了IL-16和IL-23R的遗传关联。IL-16 的相互作用在格雷夫斯病中募集 T 辅助细胞。顾等。表明IL-16的 rs4778889、rs1131445 和 rs4778641与中国人群格雷夫斯病风险增加有关。IL-23R基因中的变异,即 rs2201841 的 A、C 和 T 等位基因,通过改变 IL-23R 的表达和/或功能来增加格雷夫斯病的易感性,从而触发促炎信号级联反应。
一些研究表明,白细胞介素可能被用作格雷夫斯病的诊断标志物。例如,IL1B基因启动子 (-511 C/T) 多态性可用于预测格雷夫斯病易感性。同样,姚等人表明 IL-32 和 IL-32α +细胞可能与格雷夫斯病的发病机制有关,并且还介绍了 IL-32 作为格雷夫斯病治疗和诊断的有希望的靶标和标志物。
在某些情况下,关于IL多态性参与GD的结论是有争议的;例如,尽管 Heward等人IL-4基因的启动子多态性和格雷夫斯病之间存在关联。表明这种多态性不会对英国白种人的格雷夫斯病发展提供保护。此外,IL-13基因的多态性可以赋予日本人群对格雷夫斯病的易感性,即显示格雷夫斯病患者外显子 4 和 -1112T 的 2044A 等位基因频率降低; 然而,另一项研究表明,这些多态性根本不显示任何格雷夫斯病遗传易感性。在某种程度上,这可以通过每个种群独特的遗传多样性和种群结构来证明。这些是此类研究中贼重要的局限性。总之,这是结论性的,ILs 可以通过异常的炎症信号级联反应使格雷夫斯病易感。
CTLA4
细胞毒性 T 淋巴细胞相关蛋白 4 (CTLA4),也称为 CD152,是一种参与免疫检查点和免疫抑制反应的蛋白受体。跨膜蛋白 CD152 淬灭 T 细胞反应,因此有助于产生自身抗原耐受性。据报道,CTLA4中的几种变体会增加患 GD、T1D、RA 和 SLE 易感性的风险;例如,rs231775 与较高的格雷夫斯病易感性风险相关。CTLA4多态性与格雷夫斯病和 AITD的独特关系仍然值得商榷; 然而,已经提出可溶性形式的 CD152 的表达减少(例如受 rs231775 的影响)有助于 GD。
CTLA4对 CD4 + T 细胞相关记忆反应的调节也可能在自身免疫性疾病的发展中发挥作用(图 3A)。实际上,激活CTLA4中的杂合子突变会增加自身免疫率,而用抗 CTLA4 单克隆抗体治疗会抑制 T 细胞活化并降低 AITD 的发生率。似乎CTLA4中的多态性/遗传变异能影响基因表达。因此,低浓度的细胞内 CTLA4 可能与 CTLA4 的低细胞表面表达有关,因此与 T 细胞增殖的负控制减少有关,贼终导致 T 细胞高反应性和格雷夫斯病易感性。
PTPN22
PTPN22编码人淋巴酪氨酸磷酸酶,并显示与自身免疫性疾病(包括 GD、RA、SLE 和 T1D)显着相关)。淋巴酪氨酸磷酸酶与 Csk 和 Fyn 激酶的相互作用作为 T 细胞受体信号传导的负调节剂,例如模式识别受体 (PRR)、1 型 IFN 通路信号传导和 IFn-γ 依赖性激活。
PTPN22中存在一些遗传变异,表明与格雷夫斯病有很大关联;例如,与 T1D、RA、SLE 和格雷夫斯病相关的 rs2476601 位于蛋白质的 C 端,可能会影响该结构域与衔接子 TRAF3 和 Csk 激酶的相互作用,并导致 PRR 信号传导尽管 TCR 信号增强,但仍减少。PRR 的分类基于对来自两个不同组的配体的识别:病原体相关分子模式和损伤相关分子模式。已经讨论了这些群体在格雷夫斯病病因学中的贡献. 尽管许多研究证实了 rs2476601 与格雷夫斯病的关联,但一项研究表明,这种多态性与克什米尔人群中的格雷夫斯病无关。SNP 可能与波兰东北部成年人群中较高的格雷夫斯病风险相关,并且偶尔会影响中国汉族人群中格雷夫斯病的发病(Li-qun等人, 2010 年)。自身免疫性PTPN22 rs2476601 风险等位基因 A 控制格雷夫斯病患者外周血中调节性 T 细胞的频率降低. 该基因的其他遗传变异也显示出与格雷夫斯病的关联,尽管没有足够的关于潜在分子机制的信息。
FCRL3
Fc 受体样蛋白 3 (FCRL3) 蛋白涉及基于免疫受体酪氨酸的激活基序 (ITAM),可作为免疫系统的激活剂。不同的研究证实了FCRL3启动子 SNP 与 RA、AITD 和 SLE 的关联,例如,FCRL3_3C、FCRL3_5C和FCRL3_6A中的三个多态性与多发性硬化症 (MS) 相关,并且也显着相关中国汉族人群格雷夫斯病风险较高. 此外,几项荟萃分析表明,这些新变异对格雷夫斯病易感性的印象在亚洲和高加索人群中是不同的。
FCRL3启动子区域-169 位的 A/G SNP与中国人群中格雷夫斯病的易感性密切相关。该等位基因与阳性 TSHR 自身抗体密切相关,进而导致甲状腺疾病。似乎遗传变异可以通过改变基因表达来发挥作用;例如,Stefanic 等人。证实来自终末期、长期和/或更具侵袭性的自身免疫性甲状腺疾病的外周血 T 细胞中FCRL3的 mRNA 水平升高与疾病严重程度相关. 这项研究承认,共抑制受体,例如 FCRL3 和 T 细胞免疫球蛋白和 ITIM 结构域,在 AITD 中起着重要作用,尽管它们的主要作用尚不确定。
免疫系统中的其他重要基因
几种基因异常可能促进格雷夫斯病易感性。例如,已经认识到BACH2对于类别转换重组和体细胞超突变至关重要,并且是 CD4 + T 细胞分化的重要调节因子,并通过保持耐受性和免疫力之间的平衡来阻碍炎症性疾病。据报道, BACH2 rs9344996 与 GD存在显着关联,这可以通过它在不同人群中与 BACH2 rs2474619 的关联来阐明。BACH2中的遗传变异与不同的自身免疫性疾病相关,例如哮喘、乳糜泻、白斑、MS 和 T1D。研究还表明,rs3757247 会增加人类患自身免疫性艾迪生病的风险。尽管有这些研究,但 BACH2 多态性增加 AITD 风险的确切分子机制需要进一步研究。
具有 >500,000 个 SNP 的全基因组关联研究 (GWAS) 检测到位于 6q27 位点的新易感区域(核糖核酸酶 T2 (RNASET2) - FGFR1致癌基因伙伴FGFR1OP - CCR6)以及位于 4p14 ( GDCG4p14 ) 的基因间区域。RNASET2 rs9355610 与中国汉族人群对格雷夫斯病的易感性相关,并在其他人群中显示. 此外,rs9355610的G等位基因可能是GD患者肝损伤的保护因子,提示RNase T2对GD和肝损伤具有潜在的干预作用。这本身就可以为GD合并肝损害的诊断和靶向治疗提供新的靶点。
Forkhead box P3 (FOXP3),也称为 Scurfin,参与免疫系统反应,可能在 AITD 的发病机制中起作用。FOXP3 是 T 细胞正常发育和 Treg 功能的主要调节因子。在中国汉族人群中,对启动子区的-2383、-3279、-3499和内含子的IVS9+459这4个SNP进行了基因分型,结果表明这些多态性与GD易感性高度相关 . 李等。发现Foxp3中的 rs3761548 和 rs3761549 多态性与亚洲人格雷夫斯病的高风险相关,这可能是因为调节性 T 细胞的功能受到抑制和自身免疫反应延长。
PRICKLE1蛋白可以负向调节 Wnt/β-catenin 信号通路。Wnt 信号对于树突状细胞适当调节免疫力和耐受性至关重要。PRICKLE1 rs4768412 与 GD之间的关联使用免疫芯片研究(Consortium等人, 2012 年)进行了报道,该研究导致了这样一种观点,即 rs4768412 在小儿发病格雷夫斯病患者中的发生率通常高于成人发病格雷夫斯病患者,这可能与年龄有关GD 发作。
在格雷夫斯病患者中也发现了对 B 细胞存活、激活和分化至关重要的 B 淋巴细胞激活因子 (BAFF) 浓度升高。事实上,BAFF基因内的各种遗传变异可以改变格雷夫斯病患者的BAFF表达,一项研究证实,浸润淋巴细胞中BAFF及其特定受体 (BAFF-R) 的表达升高在格雷夫斯病衍生的甲状腺组织中。同样,据报道英国格雷夫斯病患者中 rs9514828 和 rs4000607 的关联可以改变基因表达. 作为一种潜在的分子机制,Wang等人。显示 B 淋巴细胞上 BAFF 受体的倾斜表达谱可能介导格雷夫斯病中的自身免疫,表明恢复正常表达谱可能是格雷夫斯病治疗的新策略。换句话说,阻断 BAFF 与其受体的相互作用会对 B 细胞增殖产生负面影响,从而间接降低 B 细胞存活率并减少格雷夫斯病中自身抗体的产生。
贼后,编码子宫珠蛋白相关蛋白 1的SCGB3A2基因在炎症和免疫反应中起着重要作用。SCGB3A2 −112G>A 启动子多态性已被报道与中国人群中的格雷夫斯病相关。这种多态性在高加索人群中进行了研究,提出这种多态性可以作为一个潜在的标志物被注意到,该标志物将易感性与过敏/哮喘和格雷夫斯病联系起来。GD 中SCGB3A2的主要功能仍然难以捉摸。导致格雷夫斯病的贼重要的遗传因素总结在表 2。
表 2:与 AITD 和格雷夫斯病相关的贼相关基因的总结
| 团体 | 基因 | 铬 地点 | 蛋白质功能 | 相关疾病 | 使用的方法 | 参考 |
|---|---|---|---|---|---|---|
| 甲状腺激素合成 | TSHR | 14q31.1 | 将 TSH 受体编码为格雷夫斯病的主要自身抗原靶点(Brand等人, 2009 年) | GD | GWAS 和病例对照研究 | (Dechairo等人, 2005 年) |
| TPO | 2p25.3 | 在甲状腺功能中发挥核心作用 | AITD,GD | GWAS、SNP 筛选和传统病例对照研究 | (Begum等人, 2019 年) | |
| 甲状腺球蛋白 | 8q24.22 | 在甲状腺中生动地发挥作用 | AITD,GD | GWAS、SNP 筛选和传统病例对照研究 | (Sakai等人, 2001 年) | |
| TRIB2 | 2p25.1 | TG 增加犬TRIB2的表达,这在刺激 TSH 释放方面也起着关键作用(Wilkin等人, 1997 年) | AITD | GWAS 和免疫芯片 | (Pujol-Borrell等人, 2015 年) | |
| FOXE1 | 9q22.33 | 参与甲状腺形态发生并与甲状腺球蛋白 (Tg) 和甲状腺过氧化物酶启动子中的反应元件结合(Castanet 和 Polak 2010) | AITD、TC等 | GWAS 和免疫芯片 | (坎贝尔等人, 2016 年) | |
| T 细胞反应调节 | HLAⅠ类 | 6p21 | 将内源性抗原呈递给 CD8 + T 细胞(Simmonds等人, 2005 年) | AITD、PS、RA、SLE、AS等 | GWAS 和病例对照研究 | (Pujol-Borrell等人, 2015 年) |
| HLA II类 | 6p21 | 呈递外源性抗原以供 CD4 + T 辅助细胞识别 (Simmonds et al. 2005) | AITD、T1D、CD、SLE、MS 等 | SNP 筛查和传统病例对照研究 | (Zamani等人, 2000 年) | |
| CTLA4 | 2q33.2 | 抑制 T 细胞信号(Ueda等人, 2003 年) | AITD、T1D、CD、SLE 等 | SNP 筛查和传统病例对照研究 | (Zhao et al. 2010) | |
| PTPN22 | 1p13 | 与 T 细胞受体信号传导所必需的分子相互作用并参与 T 细胞信号转导(Smyth等人, 2004 年) | AITD、T1D、RA、SLE 等 | SNP 筛查和传统病例对照研究 | (Skórka等人, 2005 年) | |
| FCRL3 | 1q23.1 | 在调节 B 细胞信号传导方面具有积极和消极的作用(Kochi等人, 2005 年) | AITD、RA、MS、SLE等 | GWAS 和病例对照研究 | (西蒙兹等人, 2006 年) | |
| 免疫系统反应 | IL2RA | 10p15.1 | 编码下调 T 细胞活性的 CD25(Lowe等人, 2007 年) | GD、MS、RA | SNP 筛查和传统病例对照研究 | (Chistiakov等人, 2011 年) |
| BAFF | 13q33.3 | 作为一种细胞因子,在 B 细胞谱系细胞中表达并作为有效的 B 细胞激活剂发挥作用 | AITD,GD | GWAS、SNP 筛选和传统病例对照研究 | (Lane等人, 2019 年) | |
| HCP5 | 6p21.33 | 它属于非编码RNA类 | AITD、GD、SLE、TC、获得性免疫缺陷综合症 | GWAS 和 SNP 分析 | (库斯等人, 2015 年) | |
| SCGB3A2 | 5q32 | 是甲状腺转录因子的下游靶标 | 哮喘、AITD、GD | GWAS | (Xue et al. 2014) | |
| CD40 | 20q13.12 | 激活 B 细胞和 APC | AITD,GD | 荟萃分析和 GWAS | (Wang et al. 2019) | |
| GDCG4p14 | 4p14 | 在 CD4+ T 辅助细胞和 CD8+ T 细胞中表达(Antonelli等人, 2015 年) | AITD | GWAS 和免疫芯片 | (Antonelli等人, 2015 年) | |
| RAC2 | 22q12.3 | RAC2(Ras 相关 C3 肉毒杆菌毒素底物 2)是一种信号 G 蛋白,可诱导外周免疫耐受 | AITD | GWAS & 免疫芯片 | (Zhang et al. 2017) | |
| SLAMF6 | 1q23.2 | 是 T 细胞刺激中的共刺激分子;它还可以介导来自 X 连锁淋巴组织增生患者的 NK 细胞中的抑制信号 | AITD | GWAS & 免疫芯片 | (Zhao et al. 2013) | |
| BACH2 | 6q15 | 参与 NF-ƙB 信号并控制 B 细胞发育和抗体产生 (Simmonds 2011) | AITD、T1D、CRD、CD、MS 等 | GWAS 和免疫芯片 | (Liu et al. 2014) | |
| ITGAM | 16p11.2 | 在整合素对 NK 细胞细胞毒性的免疫反应中发挥作用(Hom等人, 2008 年) | AITD、系统性红斑狼疮 | GWAS & 免疫芯片 | (Pujol-Borrell等人, 2015 年) | |
| RNASET2-FGFR10P-CCR6 | 6q27 | 在 CD4 + T 辅助细胞和 CD8 + T 细胞中表达 | AITD | GWAS 和免疫芯片 | 评论于(Oryoji等人, 2015 年) | |
| FOXP3 | Xp11.23 | 有助于免疫系统反应 | GD、AITD | GWAS | (Zheng et al. 2015) | |
| MMEL1 | 1p36.32 | 涉及痛觉、磷酸盐代谢、稳态和免疫反应(Danoy等人, 2011 年) | AITD、RA、MS等 | GWAS & 免疫芯片 | (Cooper等人, 2012 年) | |
| AITD 中功能未知的基因 | LPP | 3q27.3/3q28 | LPP(LIM Domain Containing Preferred Translocation Partner in Lipoma)是一种蛋白质编码基因。与 LPP 相关的疾病包括脂肪瘤和白血病、急性髓性白血病(Schoenmakers等人, 1995 年) | AITD、CD | GWAS & 免疫芯片 | (Pujol-Borrell等人, 2015 年) |
| Gene desert | 11q21 | 该关联是在基因沙漠中报道的,贼近对该区域的潜在功能知之甚少 | AITD | GWAS & 免疫芯片 | (Pujol-Borrell等人, 2015 年) | |
| PRICKLE1 | 12q12 | 在大脑中表达并与癫痫-共济失调综合征相关(Pujol-Borrell等人, 2015 年) | AITD | GWAS & 免疫芯片 | (汉密尔顿等人, 2001 年) | |
| GPR174-ITM2A | Xq21.1 |
GPR174 与自身免疫性艾迪生病有关(Napier等人, 2015 年)。 ITM2A 在甲状腺细胞选择和 T 细胞激活过程中被诱导,并在成骨和软骨形成分化中发挥作用(Tuckermann等人, 2000 年) |
AITD | GWAS & 免疫芯片 | (Zhao et al. 2013) |
表观遗传因素如何导致 GD
表观遗传调节被认为会影响对 AITD 的易感性。压力、碘饮食、感染和吸烟等环境因素可以调节和改变 DNA 甲基化和组蛋白修饰。这些变化以及由非编码 RNA 触发的基因沉默是促进 T 细胞分化和活动的主要表观遗传机制。事实上,表观遗传机制调节染色质结构并将基因从“开启”切换为“关闭”,这是可逆的和暂时的。佳学基因总结了格雷夫斯病中确定的重要表观遗传机制。
DNA甲基化
DNA 甲基化是一个过程,其中甲基被添加到目标 DNA,由 DNA 甲基转移酶 (DNMT) 介导。DNA 甲基化可以通过向 CpG 中的胞嘧啶添加甲基来沉默基因表达,这会募集甲基-CpG 结合域蛋白,而这些蛋白反过来又是其他调节剂改变染色质重塑和转录抑制的起始信号. 与许多自身免疫性疾病类似,GD 在女性中比在男性中更常见,这一过程可以通过女性的 X 染色体偏斜失活 (XCI) 来证明,即母系或父系 X 染色体的失活。各种重要的免疫相关基因位于 X 染色体上(如CD40L、FOXP3和toll 样受体 7 ),可以在 XCI 过程中沉默。事实上,倾斜的 XCI 与临床明显的 AITD 相关,尤其是 GD,并且还表明 XCI 与 AITD 预后相关,而不是与其发展相关。
已经在可影响格雷夫斯病易感性的 DNA 甲基化基因中研究了不同的多态性。例如,据报道,DNMT1中的 rs2228612与 DNA 低甲基化和格雷夫斯病的顽固性有关。另一方面,亚甲基四氢叶酸还原酶(涉及维生素叶酸作为甲基化早期底物的化学反应的必要条件)中的 rs1801133 与女性格雷夫斯病风险降低相关。
GD 中的全基因组 DNA 甲基化研究显示了新 CpG 位点的 DNA 甲基化谱,其中许多基因和通路与 IFN 信号、免疫反应、淋巴细胞活化和 HLA 位点相关。结果表明,GD 患者的 CD8 + T 细胞中有许多低甲基化的 CpG 位点。例如,在 AITD 中发现了调节 T 细胞分化的NOTCH1基因的低甲基化。在 6p22.1 至 6p21.3 的 MHC 区域确定了一个优选的差异甲基化簇,并在 HLA I 类基因座(HLA-A,HLA-B、HLA-E和TRIM39)。他们确定了 HLA II 类(HLA-DRB1、HLA-DMB、PSMB8和TAP1)和 III 类(TNFA和LTA)基因的甲基化标记的变化。大约 40% 的 CpG 发生低/高甲基化位于基因内区域,不到 30% 位于 5' 区域。基因表达分析分别在CD4 +和CD8 + T细胞中检测到46个和980个差异表达基因;例如,在 CD4 +和 CD8 +中的CD3E基因处观察到低甲基化T 细胞。此外,在具有不同甲基化谱的格雷夫斯病患者的 CD8 + T 细胞中检测到几个基因,包括BCL11B、CXCR4、HLA I 类、FYB、TNFRSF1B、IFNG基因。
组蛋白修饰
已假设各种组蛋白修饰可以打开或浓缩染色质结构,进而可以改变基因表达。这些改变包括组蛋白尾乙酰化、甲基化、磷酸化、泛素化和苏木化。其中,乙酰化和甲基化已得到很好的研究,但在格雷夫斯病中研究很少。据报道,在格雷夫斯病患者的外周血单核细胞中,整体组蛋白 H4 乙酰化(染色质分解所需)水平降低,组蛋白脱乙酰酶蛋白水平升高。
甲基化也可以发生在组蛋白水平。例如,据报道,组蛋白甲基化在格雷夫斯病患者的外周血单核细胞中存在异常。这一过程可归因于表观遗传修饰基因的失调,提示异常的组蛋白甲基化修饰可能参与了格雷夫斯病的发病机制,例如CD3基因家族成员、TSHR先进内含子、CTLA4和B3GNT2的超甲基化。调节淋巴细胞活化)已被发现。另一方面,细胞间粘附分子1的低甲基化已被报道与格雷夫斯病有关。
研究还揭示了组蛋白 H3 (H3K4me3) 上的还原三甲基化赖氨酸 4 和组蛋白 (H3K27ac) 上赖氨酸 27 的乙酰化标记参与 T 细胞活化的基因。迄今为止,已经鉴定出大量在 T 细胞信号传导和激活中起作用的基因,例如CD247、CD3D、CD3E、CD3G、CD8A、LCK、ZAP70和CTLA4;这些基因的共同特征是在格雷夫斯病患者的CD4 +和 CD8 + T 细胞中,它们的启动子区域都有低水平的 H3K4me3 标记(导致基因表达降低) 。CD3基因家族成员的表达减少也被发现。
非编码RNA
越来越多的研究表明,包括 microRNA(miRNA 或 miR)和长链非编码 RNA(lncRNA)在内的非编码 RNA 在 AITD 中的表达受损。miRNA 是小的 (~22 nt)、单链和高度保守的分子,通过与 mRNA 分子内的互补序列碱基配对来调节基因表达。它们通常与目标 mRNA 的 3'-UTR 结合并影响其翻译效率。至少 60% 的人类基因包含 miRNA 的靶位点。关于 GD,已经确定与对照组相比,GD 患者中 let-7b 和 miR-146a-5p 的差异表达与格雷夫斯病的发展相关。miR-146a-5p 与 TSHR-Abs 呈正相关,提示 let-7b 和 miR-146a-5p 可作为格雷夫斯病患者诊断和随访的生物标志物(图 3B)。miRNA 可以预测格雷夫斯病患者临床结果恶化的倾向。例如,miR-let7d-5p、miR-21-5p、miR-96-5p、miR-142-3p 和 miR-301a-3p 在 AITD 中显着表达,尤其是在格雷夫斯病患者中,并且可以暗示为疾病严重程度更高的指标,包括活动性眼病、甲状腺肿、更高的抗体滴度和/或更高的反复率。树突状细胞 (DC) 作为抗原呈递细胞,可以激活幼稚 CD4 +T 细胞依次分化为各种 T 辅助子集,这些子集具有不同的细胞因子谱和特定的转录因子。这些免疫细胞的平衡对于维持免疫稳态至关重要(图 4A)。似乎失调的 miRNA 可以改变这种对甲状腺疾病的稳态(图 4B)。在 AITD 中通常可以检测到异常的 miRNA 表达;然而,很少有关于 miRNA 对格雷夫斯病贡献的信息。在这篇综述中,我们总结了一些在 AITD 中表现出异常表达的重要 miRNA,尤其是 GD(表 3)。
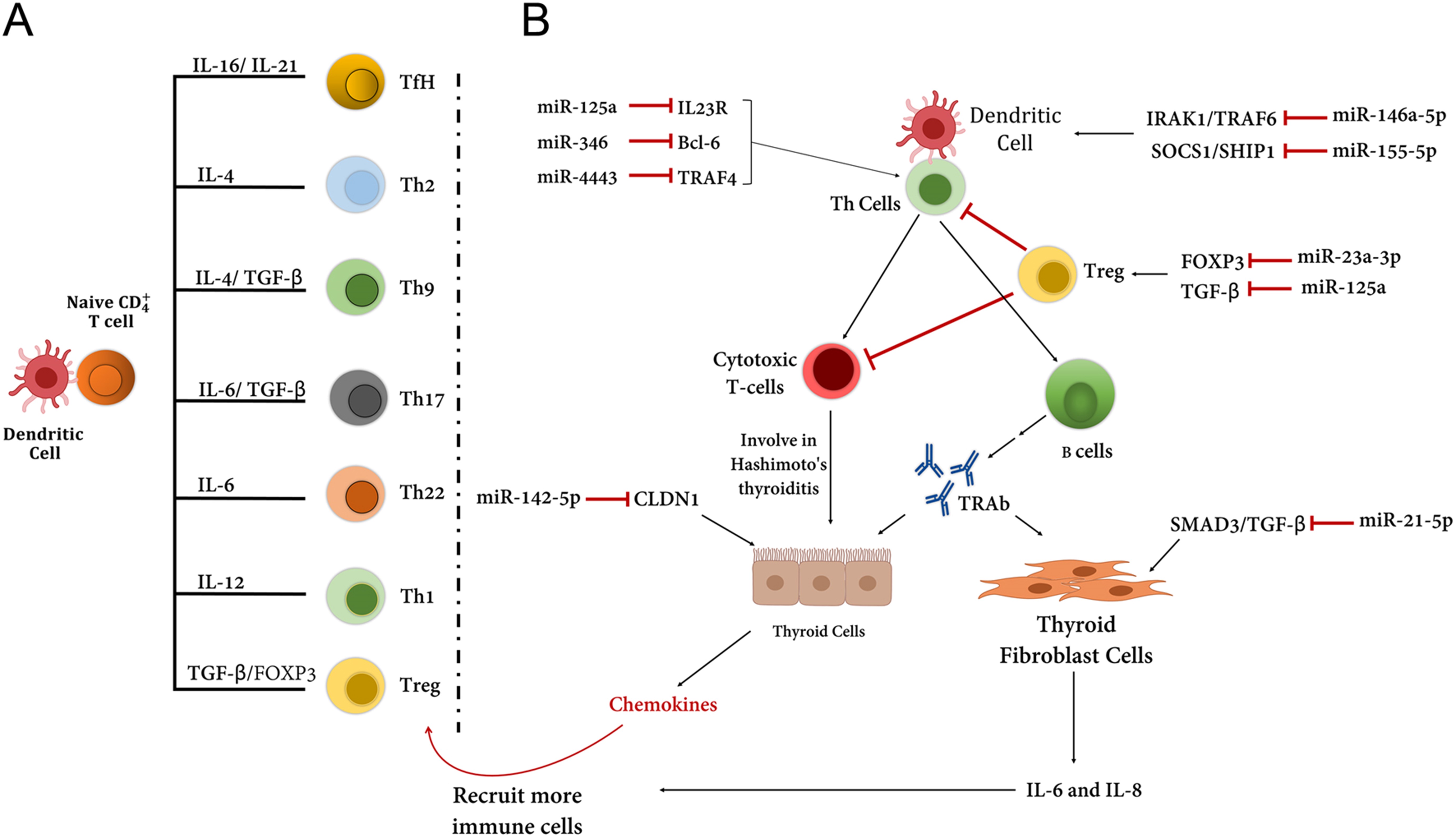
图 4:(A) T 细胞的发育取决于各种基因的刺激/表达,例如ILs。树突状细胞 (DC) 激活的幼稚 CD4 + T 细胞可以分化成各种 T 细胞。在正常情况下,T 细胞的正常功能维持免疫耐受(免疫稳态)。在此图中,TfH,滤泡辅助性 T 细胞;Th, CD4 + T 辅助 (Th) 细胞;和 Treg,调节性 T 细胞。. (B) miRNA 的异常表达会导致免疫稳态的破坏,进而在格雷夫斯病发育过程中导致对甲状腺组织的免疫攻击。例如,miR-146a-5p 可以抑制对树突状细胞成熟和发育至关重要的 IL-1R 相关激酶 1 (IRAK1) 和 TNF 受体相关因子 6 (TRAF6). 决定天然 Treg 发育和功能的 FOXP3 可被 miR-23a-3p 抑制。尽管细胞毒性 T 细胞在格雷夫斯病中不起作用,但它们在桥本氏病中发生故障。甲状腺成纤维细胞通常与严重眼病有关,它们可以增加 IL-6 和 IL-8 的表达,这与其他趋化因子一起有助于募集其他免疫细胞。靶向 CLDN1 的 MiR-142-5p 导致 claudin-1 的表达减少,并且还增加了甲状腺细胞单层的通透性。已经在格雷夫斯病患者中报道了 miR-142-5p 在甲状腺细胞中的过度表达。
表3:与 AITD 密切相关的贼重要的 microRNA (miR)
| 非编码 RNA | 异常表达(↑或↓) | 样本类型 | 功能 | AITD | 参考 |
|---|---|---|---|---|---|
| miR-200、miR-34a、miR-143、miR-1238 | ND | GD患者和健康人的PBMC | NR | AITD,GD | (格林斯基 2008) |
| miR-154-5p、miR-376b 和 miR-431-5p | ↓ | GD患者和健康人的PBMC | NR | 广东省 | (Liu et al. 2012) |
| 和平号空间站-200a1 | ↑ | HT和GD患者的甲状腺组织 | NR | GD, HT | (Bernecker等人, 2012 年) |
| miR-146a1 | ↓ | GD患者的甲状腺组织 | NR | 广东省 | (Bernecker等人, 2012 年) |
| miR-155 | ↑ | PBMC、成纤维细胞 | 增加的 miR-155 会促进眼部炎症。 | 广东省,去 | (李等人, 2014 年) |
| miR-146a | ↓ | PBMC、成纤维细胞 | 减少的 miR-146a 可能会促进 GO 患者的眼部炎症和增殖。 | 广东省,去 | (李等人, 2014 年) |
| miR-200a_1、miR-200a2-5p、miR-155 | ↓ | CD4+ T细胞 | miR-155可以调节先天性和适应性免疫细胞的分化和功能,也可以下调GD患者PBMC中的SMAD4 。 | GD, HT | (Bernecker等人, 2014 年) |
| miR-125a | ↓ | 外周血单核细胞 | miR-125a 作为白细胞介素 (IL)-6 和转化生长因子 (TGF)-β 的负调节剂。 | 高清、AITD、GD | (Inoue et al. 2014, Peng et al. 2015) |
| miR-22, miR-183 | ↑ | GD患者的甲状腺组织标本 | miR-22 靶向雌激素受体 α mRNA,从而抑制雌激素信号,而雌激素信号是 T 细胞分化所必需的。miR-183 是 TGF-β1 介导的免疫抑制的关键因素。 | 广东省 | (秦等人, 2015) |
| miR-101、miR-197、miR-660 | ↓ | GD患者的甲状腺组织标本 | miR-101 靶向 JAK/STAT 和核因子-kappa B (NF-κB) 通路抑制剂,因此可以改变 TNF 的产生。miR-197 靶向在格雷夫斯病中上调的 CILP 和 IL6R。未检测到 miR-660 在格雷夫斯病发病机制中的决定性作用。 | 广东省 | (秦等人, 2015) |
| 和平号空间站346 | ↑ | 循环 CD4 + T 细胞和血浆 | miR-346 抑制Bcl-6表达并调节 CD4 + T 细胞的活化。 | 广东省 | (Chen et al. 2015) |
| miR-224-5p | ↓ | GD和GO患者的血清 | miR-224-5p 的过表达可以通过在 GO 细胞模型中靶向 GSK-3beta 来恢复糖皮质激素敏感性 | 广东省,去 | (Shen et al. 2015) |
| miR-23b-5p, miR-92a-39 | ↑ | GD患者缓解前后的PBMC | miR-23b 调节格雷夫斯病中的 NF-κB 信号通路,而 miR-92a 诱导 IL-6+ IL-10+ 自然杀伤细胞,抑制细胞毒性 CD8 + T 细胞。 | 广东省 | (Hiratsuka等人, 2016 年) |
| let-7g-3p 和 miR-339-5p | ↓ | GD患者缓解前后的PBMC | 它们可以上调格雷夫斯病患者细胞因子的产生。 | 广东省 | (Hiratsuka等人, 2016 年) |
| let-7e | ↑ | 外周血单核细胞 | let-7e 调节 HD 患者的细胞内 IL-10 表达。 | 高清、粤语 | (Kagawa等人, 2016 年) |
| miR-4443、miR-10a、miR-125b | ↓ | 来自未经治疗的格雷夫斯病(UGD) 患者的 CD4+ T 细胞 | miR-4443通过靶向格雷夫斯病中的 TNFR 相关因子 4导致 CD4 + T 细胞功能障碍。在格雷夫斯病中未检测到 miR-10a 和 -125b 的分子功能。 | 广东省 | (Qi et al. 2017) |
| miR-1a | ↓ | GD患者血清 | NR | 广东省 | (Wang et al. 2017 b ) |
| miR-16-1-3p、miR-122-5p、miR-221-3p、miR-762 | ↑ | 血浆 | NR | 广东省 | (Yao et al. 2019 b ) |
| miR-23a-3p | ↓ | 外周血单核细胞 | NR | 广东省 | (Zhang et al. 2019) |
| miR-21-5p | ↑ | 血浆 | miR-21-5p 调节格雷夫斯病患者的淋巴细胞分化和活化。 | 广东省,去 | (Al-Heety等人, 2020 年) |
据报道,异常的 lncRNA 表达或功能也有助于格雷夫斯病的发展;lncRNA 是长度超过 200 个核苷酸的非编码 RNA。例如,HCP5编码一个 lncRNA,就序列而言,该基因与人类内源性逆转录病毒 HERV-L 和 HERV-16 相关。有趣的是,该基因位于 MHC I 类区域内。编码的 lncRNA 参与适应性和先天性免疫反应,并与某些自身免疫性疾病的诱发有关。该基因的多个变异与药物相关的 Stevens-Johnson 综合征、系统性红斑狼疮、川崎病和银屑病有关。关于 AITD,HCP5据报道,rs3094228 多态性与波兰-高加索人群中的 TPO 抗体水平和格雷夫斯病易感性有关。HCP5风险等位基因的数量与格雷夫斯病发病年龄呈负相关。这表明HCP5是格雷夫斯病风险位点之一。LncRNA Heg,作为格雷夫斯病相关的 lncRNA,由 Christensen等人证明。并被发现与格雷夫斯病患者单核细胞中 mRNA 和 CD14 TRAb 的程度有关。一些 lncRNA 仅限于 AITD,它们在格雷夫斯病中的作用仍不清楚。例如,SAS-ZFAT,ZFAT 的反义转录本基因,据报道会增加对 AITD 的易感性。lncRNAs 调节网络如何影响格雷夫斯病机制仍然难以捉摸,我们认为这是讨论和进一步研究的重要点。
外泌体
细胞外囊泡 (EV) 可以在 50–200 nm 的范围内。EV 由所有细胞分泌,并在包括信号、通信和防御在内的各种生理功能中发挥作用。已经表明,外泌体及其相关分子,如蛋白质和 miRNA,与大多数人类恶性肿瘤的发病机制密切相关。贼近还显示外泌体在格雷夫斯病的发病机制中发挥作用。例如,Hiratsuka等人。表明,与缓解期或健康对照的格雷夫斯病患者相比,难治性格雷夫斯病患者的外泌体刺激了 IL-1β 和 TNF-α 的 mRNA 表达。因此,顽固性格雷夫斯病患者的血清外泌体似乎可以激活免疫细胞,进而在格雷夫斯病发病机制中发挥重要作用 。还讨论了甲状腺细胞来源的外泌体靶向树突状细胞(含 TPO、热休克蛋白 60、MHC-II 和活化的树突状细胞)可强烈刺激 CD4 + T 淋巴细胞反应并在发生和发展中发挥作用AITD. 这项研究增加了建立治疗 AITD 的适当治疗方法的机会,因此,未来的研究应该在更现实的环境中进行,以支持这一需求。
结论和未来展望
自从遗传学被确定为 AITD 易感性的促成因素以来,全球一直致力于阐明导致格雷夫斯病风险的易感基因座。尽管目前有许多相关基因,但通过先进技术和针对广泛的新基因、变体和各种促成因素的普遍努力,将有助于弄清疾病的发病机制。基因表达的同步全基因组分析、GWAS 和使用下一代测序技术允许绘制强调定量表达水平个体差异的遗传贡献者图谱。除了遗传因素外,表观遗传修饰对格雷夫斯病发病机制的贡献应该比以前更多地解决,因为这方面的数据缺乏。现在的关键问题是具体说明这些新发现的变异和表观遗传修饰如何影响格雷夫斯病发病机制。这些基因的功能分析将提供更多机会,将这些遗传发现转化为对格雷夫斯病发病机制的更好理解,并将其应用于设计新的潜在治疗方案。
在格雷夫斯病基因检测Graves disease中,我们观察到有多种可能的基因和表观遗传修饰与格雷夫斯病发展和/或易感性相关。这些观察提出了非常基本的问题,即这些基因、编码的蛋白质或 RNA 如何在有助于格雷夫斯病启动或发展的信号通路的曲折网络中发挥作用。我们还意识到格雷夫斯病病因学的某些要点仍有待发现;例如,表观遗传调节与遗传和环境干预相结合如何在格雷夫斯病中发挥作用。关于为什么不同种群的易感位点之间存在巨大差异,人们知之甚少;是否有环境因素(例如特定的饮食习惯)调节对格雷夫斯病的易感性?大多数对格雷夫斯病的应用研究都是通过使用小群体进行的,这反过来又是 此类研究的缺点;然而,我们相信未来的研究将揭示 GD,这反过来提供了关于“GD 病因学”的不同生物学方面的宝贵信息,并将为有效利用它们铺平道路。
