












【佳学基因检测】唐氏综合征基因检测
唐氏综合征基因检测导读
21 三体综合征是指存在一条多余的 21 号染色体,导致一系列临床特征,通常称为唐氏综合征 (DS)。唐氏综合征是与人类足月生存相容的遗传最复杂的疾病之一,也是最常见的可存活常染色体非整倍体。唐氏综合征的小鼠模型涉及人类 21 号染色体的全部或部分三体性或直系同源小鼠基因组区域,为了解三重基因或基因组对唐氏综合征许多临床表现的影响提供了宝贵的见解。这项工作具有挑战性,因为 21 号染色体上有 200 多个蛋白质编码基因,它们可以对细胞、组织、器官和系统的稳态产生直接和间接的影响。虽然这种复杂性对理解唐氏综合征许多临床特征的每一个的潜在分子基础提出了巨大的挑战,但它也为加深对许多细胞类型、组织、器官和系统发育和功能的潜在遗传机制的理解提供了机会。自从首次描述 21 三体综合征以来,唐氏综合征基因解码基因检测已经了解了很多有关智力障碍和先天性心脏病遗传风险因素的知识。唐氏综合征患者实体dota2吧雷电竞 发生率较低,这支持了 21 号染色体基因的鉴定,这些基因在过度表达时可以预防dota2吧雷电竞 。阿尔茨海默病的组织病理学普遍存在,唐氏综合征患者痴呆症的患病率很高,这为了解阿尔茨海默病的病理学和治疗提供了新见解。改善唐氏综合征智力障碍的临床试验标志着一个新时代的到来,在这个时代,现在可以探索基于唐氏综合征分子病理生理学知识的治疗干预措施;这些努力为未来带来了合理的希望。
唐氏综合征基因检测的基础知识
唐氏综合征 (DS) 是智力障碍最常见的基因组疾病,是由人21 号染色体 (HSA21) 的三体性引起的。该综合征的名称来自唐氏,他于 1866 年描述了该综合征的临床表现。唐氏综合征表型涉及影响多个身体系统的疾病表现,特别是肌肉骨骼、神经和心血管系统。唐氏综合征患者通常身材矮小、肌张力低下、寰枢椎不稳定、神经元密度降低、小脑发育不全、智力障碍和先天性心脏缺陷(CHD;特别是房室间隔缺损 (AVSD))。唐氏综合征患者也更容易出现某些健康问题,包括甲状腺功能减退症、自身免疫性疾病、阻塞性睡眠呼吸暂停、癫痫、听力和视力问题、血液病(包括白血病)、反复感染、焦虑症和早发性阿尔茨海默病(AD)(图1)。其他疾病,例如大多数实体肿瘤类型,表现出逆向共病现象,在唐氏综合征患者中似乎比在一般人群中更少见。
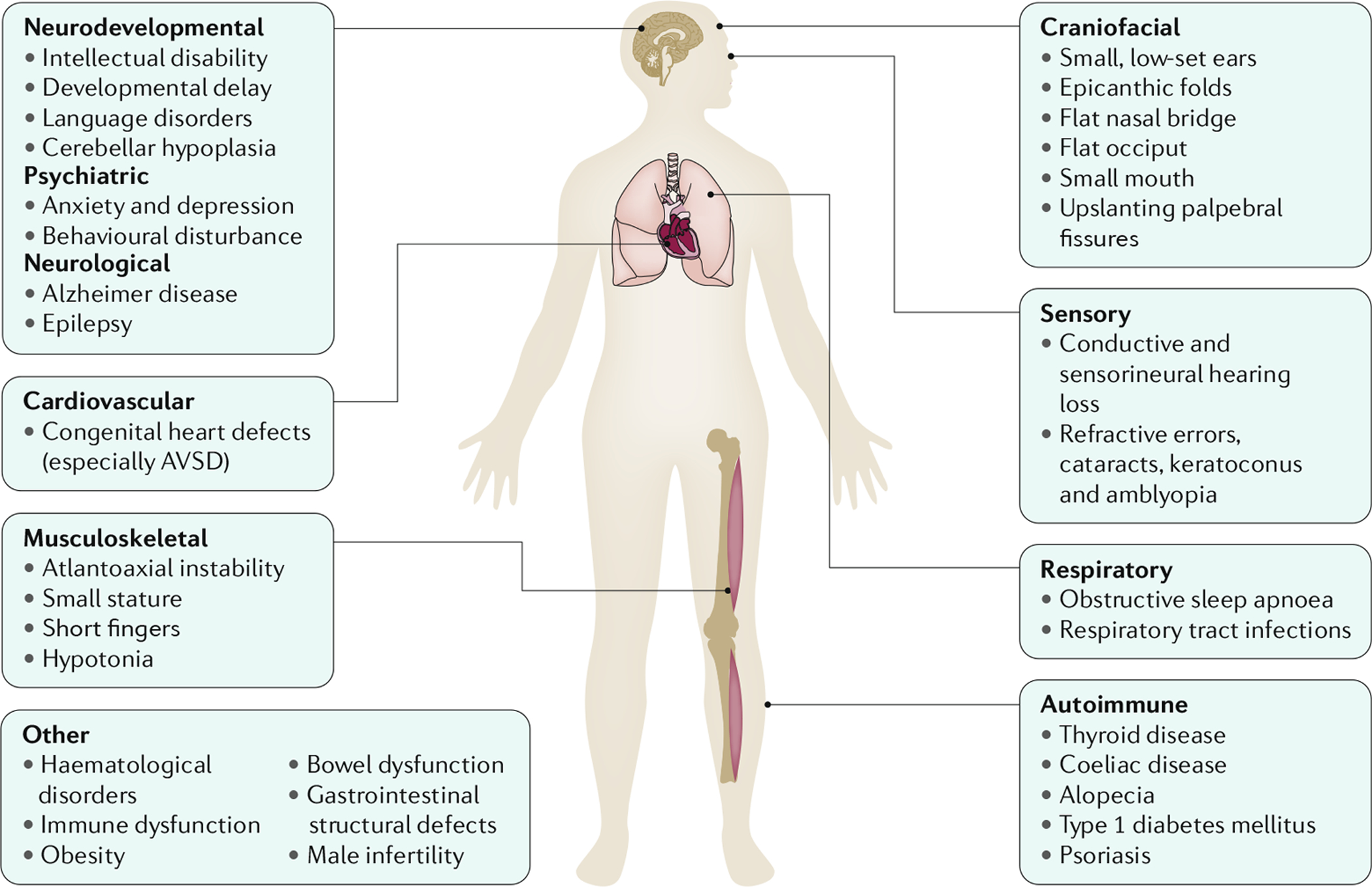
图 1: 唐氏综合症的症状和表现
患有 21 三体综合征(存在多余的 21 号染色体;也称为唐氏综合征 (DS))的个体表现出影响多个身体系统的独特症状和表现,尽管个体之间存在差异。唐氏综合征患者通常身材矮小,手指短,肌张力低下和寰枢椎不稳定。面部特征包括内眦赘皮、鼻梁和枕骨扁平、嘴巴和耳朵小以及睑裂上倾。先天性心脏缺陷很常见,尤其是房室间隔缺损 (AVSD)。与一般人群相比,唐氏综合征患者也更容易出现某些健康疾病,包括甲状腺功能减退症、阻塞性睡眠呼吸暂停、癫痫、听力和视力问题、血液系统疾病(包括白血病)、复发性感染、焦虑症和早发性阿尔茨海默病。
1959 年,人们首次报道发现了 21 号多余染色体与唐氏综合征表型之间的联系,这是遗传医学发展的重要里程碑。1990 年,人们首次开发出用于致病基因鉴定基因解码唐氏综合征的小鼠模型,2000 年,一个跨国致病基因鉴定基因解码联盟公布了 HSA21 长臂的完整核苷酸序列。在随后的 19 年里,人们在了解唐氏综合征不同表型表现的分子病理生理学方面取得了实质性进展,唐氏综合征目前被认为是一种基因表达失调疾病。此外,人们已经引入了广泛用于产前检测唐氏综合征的筛查方法。唐氏综合征患者不同症状的管理和生活质量得到了改善。然而,仍然存在巨大的挑战,包括了解该综合征每个表型成分的确切生物学机制;不同症状的治疗,包括认知功能障碍;以及唐氏综合征患者在世界各地融入社会的过程。此外,在动物模型上致病基因鉴定基因解码 200 多个蛋白质编码基因的三重化对 HSA21 的影响,更不用说三重化的非编码基因以及这些改变的下游和间接影响,这些都极具挑战性。
在致病基因鉴定基因解码指南中,唐氏综合征基因解码基因检测讨论了唐氏综合征的流行病学、目前对额外 21 号染色体导致的遗传学和病理生理学的理解,以及 21 三体综合征诊断方面的进展。此外,唐氏综合征基因解码基因检测还回顾了该综合征的治疗和唐氏综合征患者的生活质量,并展望了未来。其他一些地方已经发表了关于唐氏综合征和 21 三体综合征的评论,并提供了有关这种常见且具有挑战性的综合征的更多详细信息。
流行病学
患病率
随着全球人口的增长,唐氏综合征的终生患病率也大幅上升。例如,在美国,唐氏综合征的人口患病率从 1950 年的约 50,000 人(每 10,000 人中有 3.3 人)增加到 2013 年的约 212,000 人(每 10,000 人中有 6.7 人),这主要是由于唐氏综合征患者的儿童生存率提高 (图 2:在美国,唐氏综合征患者的预期寿命从 1950 年估计的平均 26 岁和中位数 4 岁增加到 2010 年的 53 岁和 58 岁。截至 2015 年,已报告唐氏综合征人口患病率估计值为欧洲(每 10,000 人中有 4.9 人)、欧洲(不包括前东欧集团国家)(每 10,000 人中有 6.0 人)和前东欧集团国家(每 10,000 人中有 3.3 人)。但是,除非在各国建立更多的出生登记并获得更多关于世界不同地区唐氏综合征患者历史和当前生存率的数据,否则无法可靠地计算出准确的全球估计值。
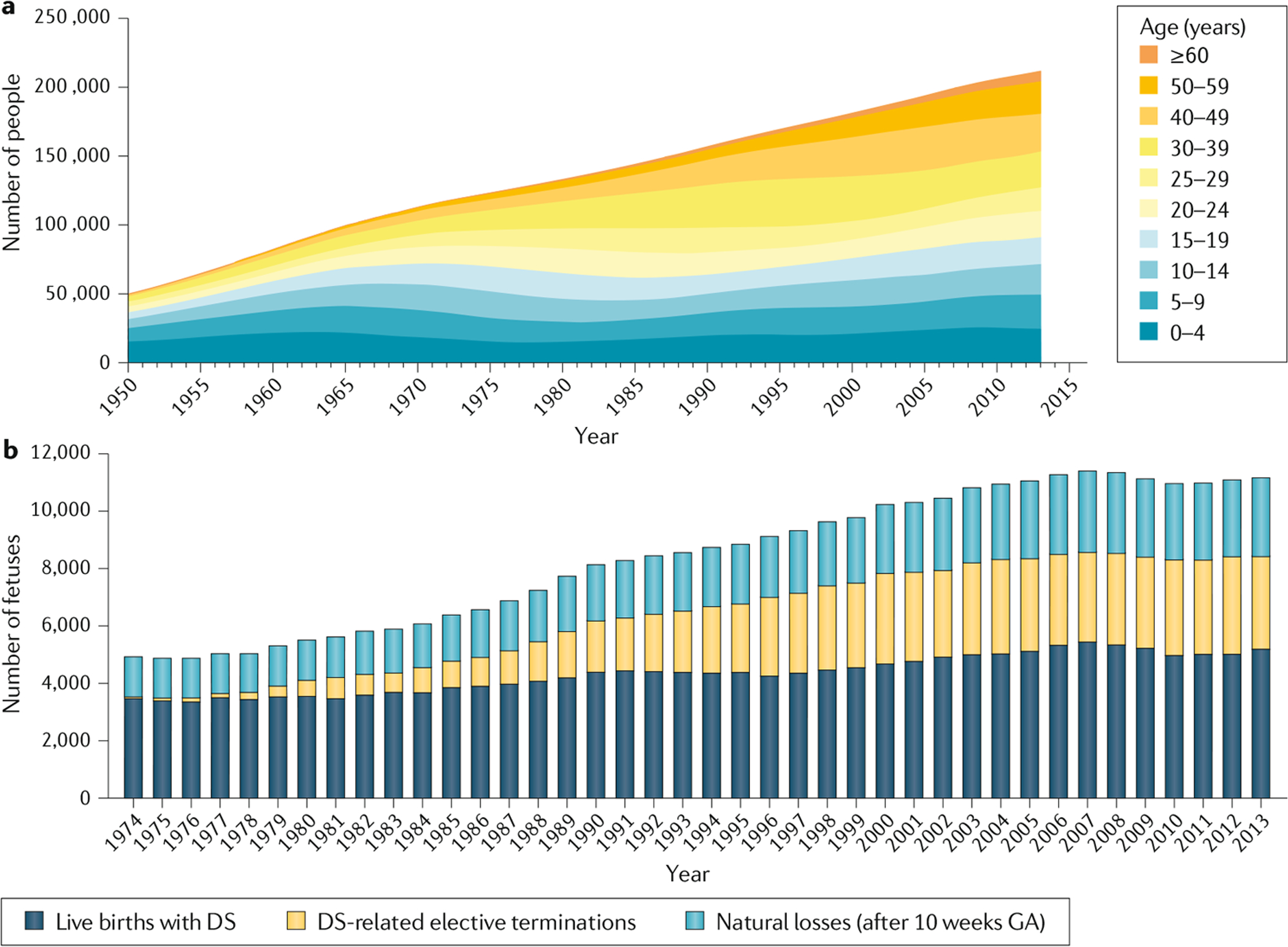
图 2 :唐氏综合征的患病率和妊娠统计
a:1950 年至 2013 年期间美国唐氏综合症 (DS) 的患病率。该图结合了美国 1950 年至 2010 年的患病率数据9和同一地区 2011 年至 2013 年未发表的患病率数据 。b:1974 年至 2013 年期间美国的妊娠结果。该图结合了美国 1974 年至 2010 年期间妊娠 10 周后怀有唐氏综合症胎儿的妇女的活产、自然流产和选择性终止妊娠的估计值17和 2011 年至 2013 年未发表的数据。GA,妊娠周龄。
唐氏综合征在所有人群中都会发生,但不同国家和不同种族的母亲受孕年龄的差异会影响活产婴儿的数量。截至 2013 年,美国未发表的数据显示,每出生 779 名婴儿中约有 1 名患有唐氏综合征(每 10,000 名活产婴儿中约有 12.8 名)(图 2a)。唐氏综合征的患病率受母亲受孕年龄的影响,不同国家的母亲受孕年龄不同,估计在妊娠 10 周时,每 365 个胎儿中就有 1 个患有唐氏综合征17(图 2b)。根据英格兰和威尔士的报告估计,其中一些妊娠会导致自然流产,孕 10 周至预产期之间流产的比例约为 32%,孕 16 周至预产期之间流产的比例约为 25%,风险取决于母亲的年龄。选择终止妊娠的数量受到每个国家筛查测试的可用性和准确性、选择产前筛查和随后进行产前检测的人数以及产前诊断为唐氏综合征后的父母的决定的影响。2013 年,美国进行了约 3,400 例唐氏综合征相关的选择性终止妊娠,导致当年出生的唐氏综合征婴儿数量减少了 33%。相比之下,2004 年澳大利亚的估计百分比下降幅度为 55%,2010 年至 2015 年期间欧洲的估计百分比下降幅度为 54%,欧洲(不包括前东欧集团国家)的百分比下降幅度为 66%,前东欧集团国家下降幅度为 32%。在中国,2003 年至 2011 年期间,唐氏综合征胎儿的终止妊娠率使总体围产期患病率下降了 55%。
来自英格兰和威尔士、斯洛文尼亚、澳大利亚和 EUROCAT 区域的致病基因鉴定基因解码表明,母亲年龄的增长抵消了产前筛查的接受度,导致从 20 世纪 90 年代至今活产中唐氏综合征的患病率保持稳定或略有下降。在欧洲,自 20 世纪 90 年代以来,活产中唐氏综合征的患病率一直在略有下降,尽管存在很大的地区差异。在南欧,1980 年至 2015 年期间活产中唐氏综合征的患病率几乎下降了一半。相比之下,在荷兰,1980 年至 2000 年期间活产患病率略有上升,但自 2005 年以来似乎一直在略有下降献25)。在美国,唐氏综合征在活产儿中的患病率从 20 世纪 80 年代开始上升,并在 2005 年后趋于平稳。自 2011 年以来,许多国家都开始提供对母体血浆中的无细胞胎盘 DNA 进行测序的服务,即无创产前筛查 (NIPS) 或无创产前检测 (NIPT);然而,还没有足够的时间去衡量监测计划对出生率的全面影响。此外,产前护理可及性的差异预计也会影响监测计划的实施。
风险因素
怀孕时高龄产妇是 21 三体综合征的主要风险因素,对于所有人类常染色体三体综合征也是如此。这种风险与卵母细胞形成过程中减数分裂期间同源染色体或染色单体不分离有关27。高龄产妇与母体减数分裂 I 和减数分裂 II 中的 HSA21 分离错误有关。此外,已观察到这些类型的母体错误的特定改变重组模式,其中只有一些与母体年龄有关。
减数分裂过程(例如重组)涉及许多基因的作用。因此,有充分的理由致病基因鉴定基因解码遗传变异是否导致人类减数分裂不分离。进行了一项包括候选基因分析和非靶向全基因组关联致病基因鉴定基因解码 (GWAS),致病基因鉴定基因解码对象为因母体错误而生下 21 三体综合征婴儿的母亲的 HSA21 不分离情况,以母亲为病例,父亲为对照。这些分析按母体 MI 或 MII 错误分层。这项致病基因鉴定基因解码的结果强调了基于错误类型的风险因素的异质性。例如,编码联会复合体组成部分的候选基因变异显示出仅限于减数分裂 I 错误的关联(例如SYCE2),而其他变异则与 MII 错误相关(例如SYCP2)。
环境因素也会影响不分离的风险,但由于确定每种因素的暴露、剂量和时间存在固有问题,因此很难识别。同样,由于病因的多样性,致病基因鉴定基因解码必须确定导致 21 三体性的错误类型(父母来源和减数分裂或有丝分裂错误的类型)。影响 21 三体性风险的环境因素包括吸烟、叶酸补充、口服避孕药和其他几种因素(致病基因鉴定基因解码环境风险因素及其局限性以及可能的暴露生物标志物(如端粒长度)的致病基因鉴定基因解码已在其他地方进行了分析)。
例如,母亲的社会经济地位 (SES) 仅与母亲减数分裂 II 错误相关 。由于 SES 是特定暴露的替代指标,一项后续致病基因鉴定基因解码将母亲职业作为一项风险因素进行致病基因鉴定基因解码,发现某些工作分类类别在唐氏综合征婴儿的母亲中比在没有染色体异常或重大出生缺陷的婴儿的母亲中更为普遍,并且与特定类型的减数分裂错误相关。初步分析显示,这些职业涉及在工作环境中接触溶剂。需要进一步进行此类致病基因鉴定基因解码,以检查工作和家庭环境中对有毒物质的特定暴露及其与母亲和父亲 HSA21 不分离的关系。接触内分泌干扰化学物质会影响减数分裂并增加非整倍体的患病率的致病基因鉴定基因解码结果强调了这一点。例如,接触普遍存在的环境污染物双酚 A (BPA) 和其他内分泌干扰化学物质会影响两性的生殖系统(包括卵巢、睾丸和生殖道)。
需要指出的是,不仅导致非整倍性的不同类型的减数分裂和有丝分裂错误很可能对特定环境暴露具有不同的敏感性,而且还必须考虑多代人(例如祖母和母亲)的暴露。例如,现在有越来越多的证据表明,在实验模型中,BPA 暴露对精子和卵母细胞具有跨代影响。
机制/病理生理学
唐氏综合征的基因检测基础
部分或完全 21 三体性(即存在部分或完全多余的 HSA21)是唐氏综合征(DS)的基因组病因。95% 的唐氏综合征(DS)患者存在游离的 21 三体性,其原因是母体减数分裂 I(~66%)或减数分裂 II(~21%);父体减数分裂 I(~3%)或减数分裂 II(5%);或合子形成后的有丝分裂(5%) 。易位导致约 5% 的受累个体出现 21 三体性,通常为t(14;21) 或 t(21;21) 46、47。约2% 的唐氏综合征(DS)患者出现 21 三体性的镶嵌现象。 21号染色体部分三体综合征较为罕见,且与一系列症状相关,这些症状随 HSA21 部分三倍体的长度而变化。
首次发表的 HSA21 序列注释了 21q 5染色体上的 225 个基因。随着对编码基因、非编码基因及调控基序元素认识的增加,HSA21 上识别的基因结构数量已显著增加。现有公共数据库中可用的基因组注释的优势和局限性已由佳学基因进行了分析。当前版本的 GENCODE/ENSEMBL(GENCODE 版本 32)列出了 233 个蛋白质编码基因、423 个非蛋白质编码基因(69 个小基因、330 个长基因和 24 个其他非编码基因)和 188 个假基因。值得注意的是,48% 的 HSA21 尚未注释,其中绝大多数包含重复元素(所有人类染色体都是如此)。
了解唐氏综合征患者对与唐氏综合征表型相关的多种表现或疾病的易感性增加的遗传病因是一个巨大的挑战。为了加大识别 21 三体综合征的具体遗传和其他影响的难度,人类唐氏综合征致病基因鉴定基因解码仅限于那些存活至足月的胎儿;在 9-14 周时确诊为唐氏综合征的产前诊断的妊娠中,约 30-40% 随后自然流产。推断所有妊娠,约 80% 的 21 三体综合征妊娠妇女在妊娠期间死亡。因此,必须认识到,在致病基因鉴定基因解码任何唐氏综合征相关疾病时,只考虑那些允许胎儿存活至足月并表现出感兴趣表型的遗传变异组合。
人们提出了两种主要假设来解释唐氏综合征表型表现背后的生物学扰动:第一,特定的 HSA21 基因剂量效应,包括过表达的 HSA21 基因的直接影响和这种过表达的下游后果;第二,发育不稳定性,其中来自额外 HSA21 的非特异性整体基因表达紊乱导致整体生物稳态的破坏。唐氏综合征相关表型的病因可能涉及这两种拟议的机制。21 三体综合征和其他三体综合征的单细胞转录组分析表明,低至平均表达的三体基因的基因剂量效应主要是由于表达这些基因的三体细胞比例较高。这导致 21 三体综合征个体各种组织中三体基因的平均表达量预计高 1.5 倍。
基因剂量效应
HSA21 三体性最简单的影响是单个 HSA21 基因剂量增加的直接影响。例如,增加APP(一种编码淀粉样蛋白前体 (APP) 的 HSA21 基因)的剂量,会增加唐氏综合征患者对早发性 AD 的易感性。虽然APP显然是一种“效应”基因,但只有APP过表达的直接和下游效应才会影响 AD 的渗透性和严重程度,还是 21 三体性的其他方面也发挥了作用,仍有待确定。值得注意的是,在 21 三体性 (Ts65Dn) 的小鼠模型中将App拷贝数恢复为两个拷贝会减轻App剂量的部分但不是全部的影响。
为了确定增加基因剂量的直接和间接后果,许多致病基因鉴定基因解码描述了 HSA21 基因的表达谱以及这些基因过表达对其余基因组的影响。这些致病基因鉴定基因解码得出了共同的发现。首先,大多数但不是全部 HSA21 基因的表达增加。确定这些不一致表达水平背后的机制非常重要,这些机制可能包括负反馈、剂量补偿和表观遗传改变。其次,在许多受影响的个体中,一些非 HSA21 基因的表达也会发生改变,这表明 21 三体性会导致下游转录和信号网络的扰动。一项荟萃分析比较了来自各种人体组织(如脑和胸腺)或细胞(如淋巴母细胞系 (LCL)、血细胞、成纤维细胞和诱导性多能干细胞 (iPSC))的 21 三体性和整倍体样本的表达谱。该分析发现,21 三体:整倍体表达比率对于失调基因通常接近 3:2,对于未受影响基因则接近 1:1。这些数据符合以下假设:效应基因(即增强或抑制基因表达的基因)在 HSA21 上的上调会导致相关下游基因表达发生类似变化,从而导致这些基因的比率发生类似改变,即 3:2 或 2:3。这种模式也有例外,基因间相互作用或其他类型的链式效应可能导致更极端的比率,从而放大最终的表达水平。例如,JAKMIP3在胸腺转录组图谱中高度上调(比率为 256.13:1),而BEX5在脑转录组图谱中高度下调(比率为 0.07:1)。
最后,正如预期的那样,21 三体细胞中 HSA21 基因和非 HSA21 基因的表达模式因所检查的组织而异。例如,干扰素反应在 21 三体成纤维细胞和 LCL 中高度上调,很可能是由于位于 HSA21 上的四种干扰素受体基因( IFNAR1、IFNAR2、INGR2和IL10RB )表达增加所致。21 三体成纤维细胞产生的 iPSC 中也存在干扰素反应增加,但在从这些 iPSC 衍生的神经元培养物中没有出现这种情况。
基因组范围的转录调控效应导致体内平衡被破坏
21 三体性也可能影响整体转录,要么直接影响HSA21 基因的转录调控功能,要么间接影响其他遗传物质的副产物。例如,至少有三个 HSA21 基因编码参与转录调控的蛋白质:ADARB1(参与腺苷到肌苷 RNA 编辑的两种蛋白质之一)、组成性剪接因子 U2AF1 和 DYRK1A(一种磷酸化剪接因子的双特异性激酶)。改变ADARB1转录水平不会改变 21 三体性 iPSC 衍生的神经元细胞中整体腺苷到肌苷的编辑水平,而在 iPSC 和由此衍生的神经元培养物中均观察到 21 三体性依赖性剪接变化。
观察发现,在三体 iPSC 和成纤维细胞中,表达发生类似改变(即上调或下调)的基因聚集在称为基因表达失调域 (GEDD) 的区域中,由此提出了遗传物质增加可能导致全基因组基因表达改变的可能性。然而,其他致病基因鉴定基因解码使用多种方法发现,没有证据表明 GEDD 专门存在于 21 三体细胞中。
使用三维荧光原位杂交 (3D-FISH) 进行的核基因组组织致病基因鉴定基因解码表明,染色体优先定位于细胞核内离散区域,称为染色体区域。三体细胞中的体内平衡可能因染色体区域的改变而被破坏。尽管对 21 三体细胞中的染色体区域及其对细胞过程的影响的致病基因鉴定基因解码才刚刚开始,但初步致病基因鉴定基因解码发现,额外的 HSA21 不会改变间期细胞核中染色体区域的整体组织66但会改变染色体压缩并将其他染色体区域从其通常的核位置移开。
21 三体综合征也会改变区域和/或整体甲基化模式,尽管甲基化变化均匀分布在所有染色体上,并且并未特异性地在 HSA21 上富集。观察到的一个模式是,与整倍体细胞相比,DS 细胞总体上倾向于高甲基化,特别是在脑样本中。此外,21 三体综合征细胞中的差异甲基化似乎定位于单个基因的离散调控区域,而不是域状区域。一项荟萃分析确定了一小部分基因,在所有检查的组织中,21 三体综合征和整倍体细胞之间的甲基化模式都不同,这表明这些改变的甲基化模式是在发育早期建立的,因此在 DS的多种组织类型中持续存在。
对于 21 三体综合征中整体改变的甲基化模式,提出了两种机制。首先,一些三重 HSA21 基因可能直接在甲基化途径中发挥作用,包括SLC19A1、FTCD、GART、CBS、PRMT2、N6AMT1、MIS18A和DNMT3L。SLC19A1、FTCD、GART和CBS都在一碳代谢途径中发挥作用,该途径是 DNA 甲基化的核心。PRMT2编码一种具有多个靶标的蛋白质精氨酸甲基转移酶,包括组蛋白70。N6AMT1编码一种负责 DNA N 6 -腺苷甲基化的甲基转移酶。MIS18A编码的蛋白质通过募集 DNA 甲基转移酶 DNMT3A 和 DNMT3B 来维持着丝粒的异染色质状态,从而抑制着丝粒卫星重复序列产生非编码转录本。DNMT3L 还与DNMT3A和 DNMT3B 相互作用并增强它们的从头甲基化活性,但本身缺乏 DNA 甲基转移酶活性。DNMT3L在唐氏综合征(DS)神经祖细胞中过表达,导致分化神经元中APP和PSD95表达增加。这些例子说明了这些 HSA21 基因的过表达可能导致唐氏综合征(DS)中的跨表观遗传变化的各种机制。
第二种假设机制是差异甲基化是由于转录因子对其结合位点的占有率改变所致。例如,RUNX1 是一种 HSA21 编码的转录因子,在 21 三体性 T 淋巴细胞中过表达,而 21 三体性细胞中的差异甲基化位点富含 RUNX1 结合基序。因此,21 三体性细胞中 RUNX1 结合位点的较高占有率会影响这些位点的 CpG 甲基化。这一假设的另一个支持来自以下观察结果:在所有检测的 21 三体性组织中,几种类型的序列基序(包括 CTCF(一种阻断增强子和启动子之间相互作用的绝缘体蛋白)的结合位点)均在差异甲基化的位点上富集。由于 CTCF 与其识别位点的结合对甲基化敏感,CTCF 占据模式可能特别受到唐氏综合征(DS)相关表观遗传扰动的影响,这可能导致细胞核内染色质的 3D 构象发生改变。
组蛋白去乙酰化和乙酰化是其他表观遗传机制,可能受某些 HSA21 基因三倍化的影响。例如,在 21 三体综合征中过表达的DYRK1A通过磷酸化和激活去乙酰化酶 SIRT1 促进组蛋白去乙酰化。此外,DYRK1A 通过与转录抑制因子 REST(也称为 NRSF) 相互作用影响染色质重塑,导致神经元基因失调,这可能导致与唐氏综合征(DS)相关的神经表型变化。
尽管基因组和转录组数据对于理解 21 三体综合征的后果及其与临床表现的个体间差异的关系非常重要,但需要与其他“组学”数据集整合才能全面了解 21 三体综合征的影响。例如,在一项蛋白质组学致病基因鉴定基因解码中,该致病基因鉴定基因解码使用一种方法分析了一对唐氏综合征同卵双胞胎的成纤维细胞的大部分蛋白质组(随后对 11 名无关的唐氏综合征患者和匹配的对照者的样本进行了跟踪调查),检测到了 HSA21 基因和非 HSA21 基因编码的蛋白质水平的广泛变化。在这两个双胞胎的转录组数据中,稳态转录本水平与蛋白质水平呈中度相关性。然而,21 三体性:整倍体基因表达和蛋白质水平的比率仅有弱相关性(Spearman 等级相关ρ = 0.34–0.51),这主要是由于非 HSA21 基因的转录本和蛋白质水平不一致。因此,大量的转录后调控在 21 三体性对不同基因表达水平的差异影响中发挥着作用。例如,对于作为与非 HSA21 编码蛋白质的异源蛋白质复合物的组成部分的 HSA21 编码蛋白质,蛋白质降解似乎可以缓冲转录水平的增加。基因富集集分析表明,与细胞周期相关的功能、细胞形态发生、脂蛋白代谢和线粒体中的细胞呼吸的蛋白质水平发生了显著改变。
在另一项致病基因鉴定基因解码80中,致病基因鉴定基因解码人员使用蛋白质组学方法主要致病基因鉴定基因解码分泌蛋白和具有细胞外结构域的蛋白81,比较了 165 名唐氏综合征(DS)患者和 98 名整倍体患者的血浆样本。在 21 三体性样本中失调的蛋白质中,许多与免疫控制、补体级联和生长因子信号传导有关。然而,失调蛋白质的身份或水平与唐氏综合征(DS)的具体临床表现之间没有明确的联系。
在 21 三体细胞(例如成纤维细胞)和器官(包括心脏)中已经观察到线粒体功能障碍的指标(它们的特性和后果已在其他地方进行了综述82 - 84)。唐氏综合征的线粒体表型包括氧化磷酸化产生的 ATP 减少、呼吸能力下降、线粒体膜电位产生受损以及线粒体结构改变。这些功能的改变导致线粒体能量代谢紊乱和氧化应激,进而可能增加唐氏综合征患者对多种疾病的易感性,包括智力障碍和 AD。线粒体功能障碍的分子基础涉及效应 HSA21 基因和关键的调节信号通路。通过对 45 项唐氏综合征(DS)基因表达致病基因鉴定基因解码85的荟萃分析,确定了 77 个持续失调的 HSA21 基因,其中NRIP1、SUMO3、DYRK1A、RCAN1、SOD1、APP和CBS直接或间接参与线粒体功能,因此是观察到的唐氏综合征(DS)相关线粒体表型的候选基因。
动物模型
唐氏综合征的动物模型,尤其是小鼠模型,对唐氏综合征的致病基因鉴定基因解码起到了推动作用。然而,致病基因鉴定基因解码这些模型时需要注意的是,唐氏综合征是一种人类疾病,无法在其他物种中精确复制。尽管如此,通过过度表达来自其他物种的 HSA21 基因组或 HSA21 基因的直系同源物,人们已经深入了解了 21 三体综合征的发育和功能后果背后的机制。关于动物模型对致病基因鉴定基因解码 21 三体综合征病理生理学的有用性的重要问题包括:哪些类型的致病基因鉴定基因解码对小鼠有指导意义,小鼠致病基因鉴定基因解码结果是否与了解 21 三体综合征的人类结果相关,以及唐氏综合征的小鼠模型是否可以用作药物测试的转化平台。
小鼠模型已经改变了唐氏综合征的基础致病基因鉴定基因解码,首先是 Ts65Dn 小鼠的开发,该小鼠含有小鼠 16 号染色体的部分三体性( MMU16) 4 , 86。在引入这种小鼠模型之前,在转基因小鼠模型中识别、克隆和表达单个基因非常困难,使用这些方法进行的致病基因鉴定基因解码得出了关于可能“导致”唐氏综合征的单个基因的广泛结论。然而,数十或数百个基因同时过度表达,由更精确定义的终端表型支持,使得对基因表达的发展、功能和全基因组扰动的致病基因鉴定基因解码有了更现实的解释。
要理解使用小鼠模型的挑战,首先必须了解 21 三体的复杂性。21 三体具有显著的表型效应,因为 HSA21 含有 200 多个蛋白质编码基因和约 400 个已知或假定重要性的额外转录本,这些转录本从受孕开始就存在于每个细胞中的额外拷贝中。相反,21 三体中的基因过表达变化很细微,与整倍体细胞相比,三体细胞的表达平均增加约 1.5 倍。单个基因的表达水平差异很大,mRNA 水平并不总是与相应蛋白质的水平相关78。对于低水平表达的基因,在许多高通量测定中无法精确可靠地测量这种差异。这种适度过表达的后果是严重的,因为多达 80% 的 21 三体妊娠者无法存活到孕15周。
小鼠表型与唐氏综合征(DS)的相关性。
所有 21 三体综合征患者均会不同程度地表现出几种唐氏综合征(DS)表型特征,包括小脑发育不全、中面部骨骼和下颌骨后缩、幼年时出现阿尔茨海默病型组织病理学以及智力障碍。DS 患者患 CHD 的风险大大增加。与一般人群相比,患有唐氏综合征(DS)的成年人患实体瘤癌症(包括乳腺癌)的总体风险较低,而患有唐氏综合征(DS)的儿童患白血病的发病率较高。所有这些特征都以某种形式存在于迄今为止生成的唐氏综合征(DS)动物模型中。例如,虽然 21 三体综合征的小鼠模型中没有出现明显的 AD 组织病理学(斑块和缠结),但据报道这些小鼠出现了 tau 蛋白的积累和内体变化。
从唐氏综合征动物模型中得出的总体结论是,给定表型的外显率 (发生率) 和表现度 (严重程度) 取决于多个基因的过度表达;唐氏综合征不是独立的单基因效应的集合。然而,许多小鼠模型 (这些模型针对不同亚群的 HSA21 基因直系同源物的三体性) 已被有效利用(尤其是与单个基因敲除相结合时,这使得特定基因在三体背景下恢复到正常的两个拷贝)来识别具有主要影响的基因,这些基因对于理解许多表型很重要 。不同的小鼠模型不是提出一个基因是否导致唐氏综合征这样的简单问题,而是提供有关特定基因或基因表达改变是否是导致表型的必要和/或充分条件的信息。因此,解释受到量化动物模型中三体性影响的能力的限制。说某个给定模型具有不完全渗透的 CHD 对于理解特定基因对特定结果的贡献没有多大用处;心室或心房隔膜发育失败、AVSD 或流出道异常都发生在不同的发育领域,涉及心脏发育不同阶段的不同细胞,并由不同的基因做出贡献。
动物模型提供的基因检测证据
HSA21 的三倍体区域或小鼠直系同源基因组区域(即小鼠染色体的片段)的范围是确定小鼠唐氏综合征(DS)模型相关性的重要考虑因素。小鼠直系同源基因组区域的 HSA21 三体性具有以下优势:这些区域受内源性转录调控,而小鼠和人类之间的启动子或增强子序列等遗传变异可能会影响转录。同样,DNA 和 RNA 中处理信号的任何正常人类特异性变异,或蛋白质中的氨基酸取代,都可能影响 HSA21 编码蛋白质和非 HSA21 编码蛋白质之间相互作用的化学计量。GWAS 和 RNA 测序的数据表明这些影响很小,但它们的潜在重要性不容忽视。相反,尽管 HSA21 和 MMU16、MMU17 和 MMU10 上的直系同源片段的基因含量相似,但它们并不相同(图 3)。在 HSA21 上预测的 233 个蛋白质编码基因中,只有 168 个在小鼠中保存良好。此外,人类和小鼠在推测功能的非编码转录本的数量和序列差异甚至更大。在对其中三种模型 Ts65Dn、Ts1Cje 和 Ts1Yey 89的比较致病基因鉴定基因解码中,强调了单个小鼠模型的局限性。
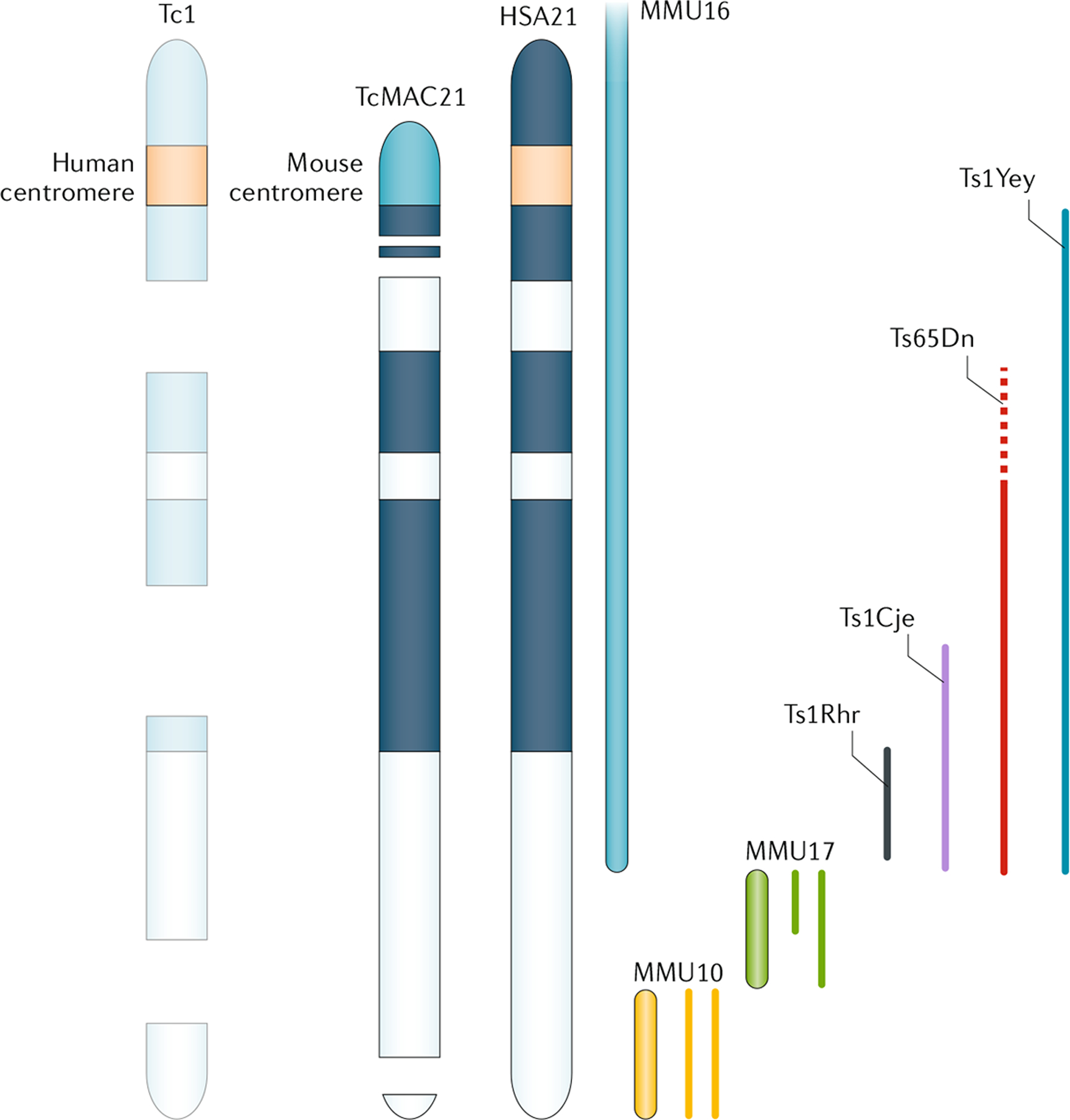
图 3 :人类 21 号染色体与小鼠染色体以及 21 三体小鼠模型的保守同源性
唐氏综合症 (DS) 是由于存在一条多余的人21 号染色体 (HSA21) 所致。目前已建立了 20 多种唐氏综合征(DS)小鼠模型,这些模型旨在过表达部分或全部 HSA21,或小鼠直系同源基因组区域。小鼠HSA21基因的直系同源物出现在小家鼠16 号染色体 (MMU16)、MMU17 和 MMU10 上。Tc1 小鼠携带突变的 HSA21,是嵌合体动物,每个个体都混合了三体和整倍体细胞,这显然是由于小鼠的人类着丝粒功能不佳所致。Ts65Dn 动物在 MMU16 上含有约 140 个基因的重复,其中一些与 HSA21 247、248 非直系同源(虚线)。 TcMAC21 动物含有 HSA21 的长臂 (HSA21q) 作为小鼠人工染色体(即带有小鼠着丝粒以确保染色体保留在每个细胞中);然而,这种人工染色体含有影响 ~8% HSA21q 基因的缺失。除 Ts65Dn 外,所有这些模型都是直接重复;也就是说,每个模型中的基因都是三体的,但它们不包含额外的染色体或着丝粒。本文讨论了 Ts1Rhr、Ts1Cje、Ts1Yey 和 Ts65Dn 小鼠模型。
过去并未强调这些差异,因为仅存在一种含有自由分离的 HSA21 的小鼠模型(Tc1)。迄今为止,Ts65Dn 是已报道的唯一一种分离额外小鼠染色体的唐氏综合征(DS)小鼠模型;所有其他模型都是通过直接复制与 HSA21 直系同源的小鼠染色体片段而制成的。Tc1 小鼠的基因组测序发现 HSA21 存在大量缺失、突变和重复,导致这些小鼠中 >20% 的 HSA21 基因表达受损。然而,Tc1 最大的缺点是相当大比例的细胞中整个 HSA21 会自发丢失,导致镶嵌性三体性(即每只动物都是三体和整倍体细胞的独特镶嵌体)。由于发育是由细胞间通讯驱动和调节的,如果邻近细胞突然改变倍性,个体的许多方面都可能发生变化。由于 HSA21 的丢失是随机的,因此每只动物的发育都是独一无二的。因此,这些重排和嵌合现象需要谨慎解读使用 Tc1 小鼠的致病基因鉴定基因解码。一种新的模型含有 HSA21 长臂上 92% 的蛋白质编码基因作为小鼠人工染色体,避免了嵌合现象的问题,并且具有在以前的小鼠模型中观察到的 21 三体综合征的许多表现。
证明使用 Cre–Lox 系统可以实现定向染色体间重组,为创建用于剂量不平衡致病基因鉴定基因解码的新小鼠模型铺平了道路。迄今为止,已创建了 20 多个小鼠模型,这些模型是 MMU16、MMU17 和 MMU10 的不同 HSA21 同源部分的三体7 (图 3)。一组三个小鼠品系共同包含与 HSA21 直系同源的所有小鼠基因组区域的完整重复已被描述。理论上,这些小鼠品系的三向杂交应在八分之一的后代中产生“完全三体性”。虽然这种低频率仅在某些应用中是可以接受的,但在实践中,完全三体性的频率远低于预测的孟德尔频率,使得 Tc1 或 MAC21 小鼠等模型成为最大限度基因表现的最可行选择。
95% 的唐氏综合征患者有额外的自由分离的 HSA21,因此有额外的着丝粒和端粒,而唐氏综合征的片段重复小鼠模型却没有。在这些模型中,三体性通过直接的染色体内重复发生,因此染色体(和着丝粒)的数量保持不变。哺乳动物中额外的着丝粒的一个影响是男性生育能力下降,这在患有唐氏综合征的人类男性和唐氏综合征的小鼠模型中都有观察到。其确切机制尚不十分清楚 。此外,与整倍体细胞相比,三体细胞似乎增殖更慢和/或细胞周期延长。尽管这些致病基因鉴定基因解码的许多结果相互矛盾,但细胞周期的微小变化都可能对发育产生重大影响。由于人类发育需要43次群体倍增才能从单细胞进展到胎儿中估计的一万亿个细胞(如果所有细胞都存活下来——实际数量显然要高得多),细胞周期长度的微小增加都可能导致可识别的表型,例如身材小巧、手指短、骨骼后退、脑神经元密度降低和细胞组织减少。
从动物模型中获得的病理生理学见解
在生物医学致病基因鉴定基因解码中,小鼠模型提供了一种重要的转化工具,可用于识别和优先考虑治疗方法以减轻疾病的影响。为了将小鼠中唐氏综合征(DS)样表型与特定基因(和基因组)的三体性联系起来,人们已经开发出 20 多种具有部分7 号染色体三体的小鼠模型(图 3)。尽管小鼠为了解唐氏综合征的致病机制提供了良好的哺乳动物遗传模型,但重要的是要记住小鼠不会患唐氏综合征。然而,小鼠模型确实说明了基因对三体性影响的几个基本原理,并且临床前药理学改善似乎很有希望,尤其是对认知影响。导致有害表型的三体基因是药物开发以改善唐氏综合征特征的目标。为了说明小鼠模型的应用,讨论了使用小鼠模型来确定三体性对脑功能、心脏发育和癌症风险的影响的遗传基础的例子。
许多临床前致病基因鉴定基因解码强调改善认知功能,这可能是药物干预最困难的目标。Ts65Dn 和许多后续模型表现出学习和记忆缺陷以及与海马突触功能改变一致的电生理变化。有趣的是,在标准记忆测试中,Ts65Dn 小鼠与 Ts1Cje 小鼠相比表现出明显的学习缺陷。Ts65Dn的三体基因数量比 Ts1Cje 多(图 3),其中一个是APP。通过将APP的无效等位基因杂交到 Ts65Dn 背景中,将APP拷贝数恢复为两个,大大提高了这些小鼠的表现,尽管没有达到整倍体小鼠的水平。
~50% 的唐氏综合征婴儿患有 CHD,小鼠模型也显示出一些类似的发育缺陷,尽管发生率低于人类婴儿。为了确定当基因以三个拷贝存在时会导致 CHD,致病基因鉴定基因解码了与 HSA21 直系同源的 MMU16 中~23 Mb 区域的七个 (部分或完全) 三体性,从而将导致 CHD (特别是类似于唐氏综合征的间隔缺损) 的区域缩小到 MMU16 的 4.9 Mb 部分。然而,细分 4.9 MB 区段的三个较小的三体性没有心脏缺陷,这表明至少有两个基因是三体的才会导致 CHD。用不同的方法,致病基因鉴定基因解码了 CHD 风险的三体性和二体性修饰因子之间的相互作用。将Creld1 (一种与家族性 AVSD 有关的基因)半合子小鼠与 Ts65Dn 和 Ts1Cje 小鼠杂交。尽管Creld1 +/−小鼠的心脏发育正常,但Creld1 +/− Ts65Dn 小鼠的室间隔缺损发病率显著增加,但Creld1 +/− Ts1Cje 小鼠的室间隔缺损发病率却没有增加。在 Ts65Dn 中为三体而 Ts1Cje 中不为三体的基因中,有 14 个在心脏发育过程中表达,包括连接粘附分子 2 ( Jam2 ),当它在斑马鱼中过表达时,会持续增加心脏异常的发生率 。在Creld1 +/− Ts65Dn 小鼠中恢复两个Jam2拷贝可阻止这些小鼠 CHD 发病率的增加。因此,Jam2的三体性是二体风险因子基因Creld1影响心脏发育的必要条件。
动物模型对于了解唐氏综合征患者的癌症风险也发挥了重要作用。流行病学致病基因鉴定基因解码提供了强有力的证据,表明唐氏综合征成人患者多种癌症的发病率降低。重要的是,唐氏综合征小鼠模型的致病基因鉴定基因解码证实了这些结果,表明部分 21 三体性可预防多种癌症。例如,家族性腺瘤性息肉病 (FAP) 是一种先天性疾病,涉及在早期形成癌前腺瘤性息肉(主要在结肠中),是由肿瘤抑制基因腺瘤性息肉结肠 ( APC ) 的突变引起的。有趣的是,携带FAP 患者体内发现的Apc突变 ( Apc Min )的 Ts65Dn 小鼠比整倍体小鼠患腺瘤性息肉的可能性低 50% 。 Ts1Rhr 小鼠在 Ts65Dn 中三倍体基因中只有 33 个是三体的,腺瘤性息肉形成率也有类似的降低。将这 33 个基因中的一个(原癌基因Ets2)恢复为两个拷贝,这种减少就会大大逆转。此外,这 33 个基因(包括Ets2 )单体化的Apc Min小鼠比整倍体Apc Min小鼠患肿瘤的可能性更大。因此,对三体性和单体性小鼠模型的评估揭示了Ets2过表达的保护作用和Ets2表达降低的肿瘤发生允许作用。
临床前致病基因鉴定基因解码和药物开发
基于对唐氏综合征小鼠模型的临床前观察,已开展了多项旨在改善唐氏综合征患者认知能力的药物和营养剂临床试验(例如,利凡斯的明的NCT00748007、 RG1662 的NCT02484703和NCT02024789以及美金刚的NCT01112683)。虽然对所有这些试验的深入讨论超出了本《入门指南》的范围,但为了便于说明,唐氏综合征基因解码基因检测讨论了导致 RG1662(也称为 basmisanil 或 RO5186582)I 期和 II 期试验的临床前致病基因鉴定基因解码,RG1662 是 GABA A α5 受体的反向激动剂。 Ts65Dn 小鼠的认知测试表明,这些小鼠在依赖海马功能的任务中存在学习和记忆缺陷,这促使电生理学致病基因鉴定基因解码表明 Ts65Dn 小鼠海马切片中的长期增强 (LTP) 降低。 LTP 降低源于过量的 GABA 介导的抑制,可以通过使用 GABA A抑制剂戊四氮 (PTZ) 治疗来逆转,恢复抑制-兴奋平衡,从而提高认知测试的表现,即使在年轻成年小鼠中也是如此。
这些致病基因鉴定基因解码彻底改变了人们对唐氏综合征认知功能障碍及其治疗的认识,它们不仅表明唐氏综合征认知缺陷是可以治疗的,而且表明这种治疗对成年人也有效,而此前人们普遍认为认知改善只能在生命早期的短时间内实现。此外,这种治疗具有持久的效果,因为在停止 PTZ 治疗 3 个月后仍测量到 LTP 有所改善 。因此,治疗认知缺陷并不是一个遥不可及的模糊目标,而可以而且应该立即着手解决。GABA A α5 受体的反向激动剂可恢复Ts65Dn小鼠的 LTP 并提高其认知测试成绩。 2013 年结束的 I 期试验 ( NCT01436955 )成功评估了 RG1662 对唐氏综合征患者的安全性和耐受性。然而,2014 年开始的 II 期试验于 2016 年提前停止,因为 RG1662 没有显示出疗效。
尽管这项特定试验设计缺乏疗效令人失望,但该试验代表了几项进步。首先,它表明制药行业现在有信心治疗智力障碍,这对唐氏综合征患者群体来说尚属首次。其次,能够在美国和欧洲招募到能够进行试验中使用的复杂测量的多个中心,再加上唐氏综合征患者群体在招募试验人员方面的大力支持,这清楚地表明了致病基因鉴定基因解码人员、临床医生和唐氏综合征患者的兴趣。最后,应用专门为唐氏综合征设计的认知测试是试验的重要组成部分。虽然这项试验的官方结果摘要尚未公布,但参与试验的许多致病基因鉴定基因解码人员已经报告了治疗唐氏综合征患者的原则。总体而言,试验结果令人失望,但人们对未来改进治疗和试验设计方法抱有乐观态度,并为进一步的试验提供了信息,例如最近的 II 期试验,该试验使用含有表没食子儿茶素-3-没食子酸酯 (EGCG) 的绿茶提取物来改善认知结果。
阿尔茨海默病的机制
几乎所有患有唐氏综合症的成年人在 40 岁之前都会出现类似 AD 的神经病理学。AD 的特点是有一个漫长的无症状临床前阶段,在此期间,淀粉样蛋白病理学会出现,大约在观察到任何认知障碍之前 15-20 年。这种临床前阶段存在于常染色体显性遗传的 AD和与唐氏综合症相关的早发性 AD。有充分证据表明, 21 三体综合征患者中APP剂量的增加是唐氏综合症患者患 AD 风险增加的必要条件,尽管将APP剂量与神经退行性病变联系起来的潜在机制尚不清楚。然而,至少有三种分子机制被提出来解释唐氏综合症患者患 AD 风险的增加(图 4)。
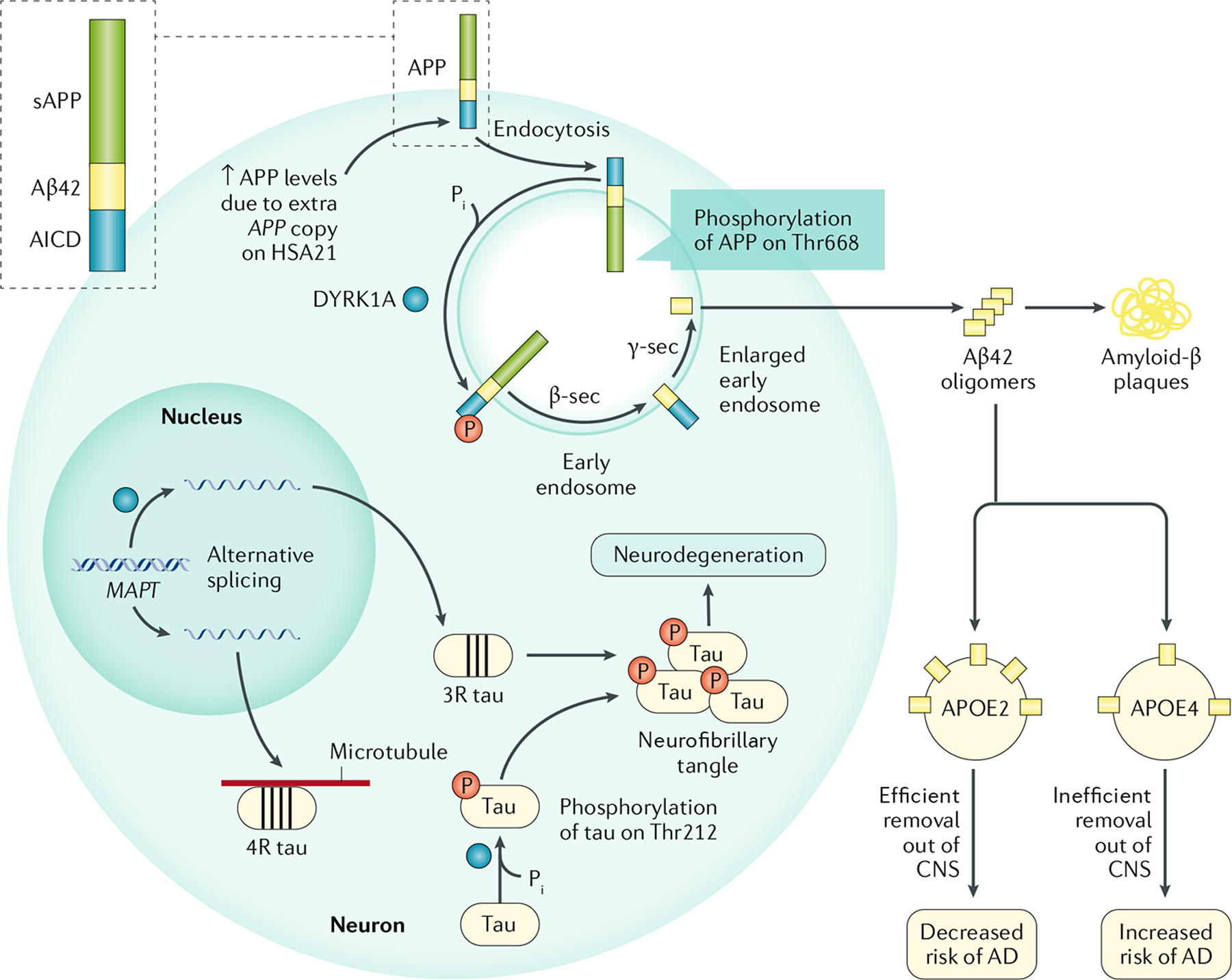
图 4:唐氏综合症患者为什么具有阿尔茨海默病高风险
患有唐氏综合征 (DS;21 三体综合征) 的个体中,人类21 号染色体 (HSA21)上APP(编码淀粉样蛋白前体 (APP))和DYRK1A(编码双特异性酪氨酸磷酸化调节激酶 1A (DYRK1A))基因剂量的增加,增加了他们患阿尔茨海默病 (AD) 的风险。APP 可经受非淀粉样变性加工(未显示)和淀粉样变性加工,由 β-分泌酶 1 (β-sec) 和 γ-分泌酶 (γ-sec) 产生神经毒性 Aβ42 肽。APP 水平升高会导致 Aβ42 水平升高,而 DYRK1A 对 APP 在 Thr668 位点的磷酸化会增加 APP 的淀粉样变性加工。DYRK1A 在细胞中还有其他靶标,包括剪接因子和微管结合蛋白 tau。剪接因子的 DYRK1A 磷酸化会改变MAPT mRNA(编码 tau)的剪接,导致含有三个微管结合结构域(三重复 (3R) tau)的 tau 水平升高,而 4R tau 水平降低。这种不平衡会导致神经原纤维缠结 (NFT) 的形成,可能是因为 3R tau 对微管的结合亲和力较低。此外,tau 中 Thr212 的 DYRK1A 磷酸化会改变蛋白质的构象,降低 tau 对微管的亲和力并导致 NFT 形成。APOE 的 ε4 等位基因(APOE ⋆ ε4)的存在会改变Aβ 249的加工、沉积和清除,因此是 AD 的主要风险等位基因。AICD,APP 胞内结构域;APOE,载脂蛋白 E;CNS,中枢神经系统; P i,无机磷酸盐;sAPP,可溶性淀粉样蛋白前体。
APP 和 A β 42
由于存在额外的APP基因拷贝而导致的APP表达增加与早发性 AD 密切相关。APP基因座的基因组重复是一些家族性早发性 AD 病例的原因 。在患有完全 HSA21 三体的唐氏综合症患者中,位于 HSA21的APP的额外拷贝导致脑中 APP 及其裂解产物(包括神经毒性 Aβ42 肽)的水平升高。然而,患有 HSA21 部分三体性且 APP 为二体的个体不会患上早发性 AD,并且具有正常的 APP 表达水平。
DYRK1A
第二种机制涉及DYRK1A过表达的多种神经病理学影响。HSA21 编码的 DYRK1A 在 Thr668 位点磷酸化 APP,导致过表达人类DYRK1A基因的唐氏综合征小鼠模型细胞中的磷酸化 APP 水平升高。这种磷酸化促进了 β-分泌酶和 γ-分泌酶对 APP 的淀粉样变性加工,导致神经毒性 Aβ42 肽的产生增加。这些小鼠大脑中的磷酸化 APP 和 Aβ42 水平升高,而唐氏综合征患者大脑中的磷酸化 APP、DYRK1A 和 Aβ42 水平升高。
DYRK1A 还对与 AD 病理生理学有关的另一种分子,即微管稳定蛋白 tau 有影响。DYRK1A 在 Thr212 位点磷酸化 tau,该残基在 AD 患者中被过度磷酸化。tau 的异常过度磷酸化会改变其构象,导致对微管的亲和力降低,并导致微管不稳定、神经原纤维缠结 (NFT) 形成和细胞死亡 。此外,DYRK1A 还会磷酸化剪接因子,从而改变 tau 编码基因MAPT的 mRNA 剪接。MAPT 的可变剪接会导致产生具有三个或四个微管结合域的 tau 蛋白异构体(分别为三重复 (3R) tau 和 4R tau)。维持正常脑功能需要 3R tau 和 4R tau 保持同等水平,在散发性 tau 病中检测到了两者水平的不平衡。DYRK1A 过表达会增加 3R tau 的丰度,而 4R tau 则会减少,这被认为会导致 NFT 的形成,因为 3R tau 对微管的亲和力低于 4R tau。
内体功能障碍
APP的额外拷贝足以通过 Aβ 非依赖机制引起内体增大和细胞内运输缺陷。RAB5 介导细胞表面蛋白的内吞作用和早期内体的同型融合。RAB5 阳性早期内体增大是一种与 RAB5 过度激活相一致的表型,存在于散发性或家族性 AD 患者脑内的神经元中以及患有 AD 的唐氏综合症患者。事实上,循环外周血单核细胞中的内体大小可能作为 AD 相关内体病理的血液生物标志物。
唐氏综合征中的其他因素也会影响内体功能。APOE 的 ε4 等位基因(APOE ⋆ ε4)是晚发型 AD 最重要的遗传风险因素,因为 ~80% 的 AD 患者至少有一个APOE ⋆ ε4等位基因。APOE ⋆ ε4还被认为会增加唐氏综合征老年人患痴呆症的风险,尽管程度低于整倍体个体中散发性 AD 的风险。三体神经元的内体增大被认为会导致轴突运输缺陷,从而导致神经元变性。此外,APOE 调节淀粉样蛋白原纤维的内体运输 ,携带APOE ⋆ ε4的小鼠脑中存在早期内体增大。APP基因剂量、Aβ42 和 APP 的 C 末端片段 C99(也称为 βCTF)在内体功能障碍的发展中也发挥着重要作用。C99 水平升高是唯一与内体异常增大和增殖相关的 APP 相关改变,这种改变在所有形式的 AD 中都很常见。
由于APP基因剂量是唐氏综合征患者罹患 AD 风险的重要决定因素,因此各种降低唐氏综合征患者 APP 水平的疗法可能对治疗 AD 有效,包括降低APP表达(使用反义方法)和 APP 产生(使用翻译抑制剂如 Posiphen)、阻断 APP 裂解(例如通过 BACE 或 DYRK1A 抑制或使用 γ-分泌酶调节剂)或去除 Aβ(通过主动或被动 Aβ 免疫)。
诊断、筛查和预防
产前筛查
在发达国家,基于实验室的唐氏综合征产前筛查是常规产前保健的一部分。筛查是识别高风险妊娠的一种方法,从而限制诊断程序,将医源性流产的风险降至最低。自 20 世纪 80 年代以来,主要的产前筛查方法结合了测量母体血清生化分析物,最近又测量了妊娠前三个月胎儿颈部透明带 (NT;颈后一囊液体) 的大小。最初,测量了妊娠中期母体血清和羊水中的甲胎蛋白水平,如果甲胎蛋白水平为正常妊娠的 ~70% 则表明胎儿罹患唐氏综合征的风险较高。最近,已经测量了更多的孕妇血清分析物,包括β-人绒毛膜促性腺激素、未结合雌三醇、抑制素A和妊娠相关血浆蛋白A。所有这些标志物的水平都会随着孕龄的变化而变化,有些标志物在妊娠早期能更好地区分21三体综合征和整倍体胎儿,而有些则在妊娠中期更能区分。因此,需要通过超声检查正确确定妊娠日期,以便准确解释血清分析物测试结果。对于每位女性,胎儿患唐氏综合征的风险是使用计算机算法计算出来的,输入原始分析物值、孕龄和人口统计信息,如母亲年龄、地理民族背景、吸烟状况以及是否患有糖尿病。临床实践中使用的数值风险截断值的参考点不同(即受影响胎儿的风险与活产婴儿的风险)。生下活产婴儿患有唐氏综合征的几率低于在妊娠中期生下胎儿患有唐氏综合征的几率,因为有些胎儿会在妊娠中期自然流产18。专业指南建议对筛查结果呈阳性的孕妇进行检测后咨询和诊断测试,例如羊膜穿刺术或绒毛取样,然后进行基因分析。
20 世纪 80 年代末和 90 年代初,产前超声检查被纳入常规护理,并且一些异常被证实与唐氏综合征(DS)有关。值得注意的是,胎儿解剖学发现并不能诊断 DS,事实上,很多患有唐氏综合征(DS)的新生儿的产前超声检查结果看起来是正常的。可能表明唐氏综合征(DS)的孕早期超声检查特征包括相对于胎龄的 NT 测量值增加以及其他四个孕早期标志物:鼻骨缺失、额颌角增大、三尖瓣反流以及静脉导管缺失或血流减少。孕中期异常扫描已成为孕 18 至 20 周的常规检查,包括可量化的标志物,如颈部皮褶增厚以及股骨和肱骨长度测量值。其他所谓的“软”超声标记包括囊性水瘤、突出的舌头、脉络丛囊肿、轻度脑室扩大、心脏缺陷、肠道回声增强、十二指肠闭锁、肾盂扩张、双侧第五指弯曲指畸形和大脚趾和第二脚趾之间的宽间隙。
结合使用妊娠早期或中期母体血清分析物定量和 NT 测量,阳性筛查结果的阳性预测值 (PPV) 约为 3-5% 。由于这些 PPV 较低,人们长期以来一直对更精确的 NIPS 胎儿染色体非整倍体感兴趣。2011 年,母体血清中游离 DNA 测序开始在临床上使用。使用大规模并行测序,可以计数 DNA 片段,将其映射到基因组的特定区域,然后与参考值进行比较;映射到 HSA21 的片段数量过多提示(但不能诊断)DS。在很短的时间内,NIPS 已成为基因组医学临床实施最多的例子,迄今为止已进行了约 1000 万次测试。目前,在美国,胎儿患唐氏综合征风险较高的孕妇(孕妇年龄 >35 岁、家族史阳性、血清分析物和/或 NT 测量值阳性、提示超声异常)会接受 NIPS 作为主要筛查。在欧洲许多国家,NIPS 开始作为主要筛查阳性后的二次检测。目前,只有比利时和荷兰为所有孕妇提供 NIPS 作为主要筛查,无论其先验风险如何。
一项荟萃分析检查了 NIPS 在生下 DS胎儿风险高或低的孕妇中的 PPV 。对于 DS,高风险和低风险孕妇的 PPV 分别为 91% 和 82%。直接比较同一孕妇中分析物和/或 NT 测量与 NIPS 的预测性能的致病基因鉴定基因解码已证明使用测序可使 PPV 增加10-20倍。重要的是,NIPS 不依赖于孕周,可以在妊娠 10 周和分娩之间的任何时间进行。由于 NIPS 的表现优于目前的护理标准,因此在一些拥有国有医疗系统的国家,正在转向将 NIPS 作为 DS的主要筛查。
由于 NIPS 是一种非侵入性检查且具有非常好的 PPV,人们一直担心这种检查会对唐氏综合征婴儿的活产率产生何种影响。一项初步致病基因鉴定基因解码发现,在 8 个国家/地区推出 NIPS 之前和之后,唐氏综合征婴儿的活产数量没有差异。但是,如果无法获得所有产前检测结果,则不清楚在此期间会有多少唐氏综合征婴儿出生。需要进行更广泛的致病基因鉴定基因解码来评估 NIPS 对唐氏综合征发病率的影响。尽管许多女性选择不进行产前检测,因为这不会影响她们的生育决定,但一项致病基因鉴定基因解码表明,产前知道自己的婴儿患有唐氏综合征的女性在心理测试中的结果比出生时发现婴儿患有唐氏综合征的女性要好。据报道,产前诊断唐氏综合征有多种好处,包括为父母提供教育机会、在分娩前与儿科专科医生会面以及改变分娩地点以便有合格的儿科专科医生提供护理,从而使母亲和婴儿能够留在同一家医院。准确的产前筛查未来的好处可能包括有机会在胎儿期开始治疗以改善认知能力(稍后讨论)。
管理
每个唐氏综合征患者都有其独特的优势和挑战,这些优势和挑战会在其一生中发生变化。有些人从出生起就需要大量医疗投入,而有些人可能很少有健康并发症。同样,有些人整个成年期都需要社会照顾和支持,而有些人则能够独立生活。唐氏综合征患者的一些健康问题比一般人群更常见,包括先天性心脏病、阻塞性睡眠呼吸暂停、甲状腺疾病、痴呆、癫痫、胃肠道疾病、听力和视力问题、智力障碍和发育障碍、精神疾病、免疫功能障碍、血液系统疾病和肌肉骨骼问题。应定期筛查这些表现,并且已有针对唐氏综合征儿童的共识筛查指南,但尚未制定针对成人的指南。针对成人的服务通常比针对儿童的服务更专业,并且护理通常需要由多个不同的医疗团队管理。目前尚未就谁应该监督唐氏综合征成人患者的护理达成共识,但初级保健医生通常会承担这一角色;有时,儿科医生可能会一直参与到成年早期,在某些国家,例如英国,智力障碍精神科医生最有可能在社区团队中提供医疗支持。由于缺乏共识,唐氏综合征成人患者可能会错过定期筛查和主动治疗,只有在临床上出现明显困难时才会进行干预。
唐氏综合征患者的疾病表现可能有所不同。存在诊断掩盖和症状错误归因的风险,而唐氏综合征患者沟通困难的普遍性又会加剧这种情况。由于唐氏综合征涉及多个系统,因此护理需要来自一系列医疗、社会护理和教育团队的多学科投入。
围产期管理
怀有确诊为唐氏综合症的胎儿的孕妇需要在整个围产期接受定期监测和支持。怀有唐氏综合症胎儿的孕妇流产风险较高,估计 12 周后胎儿自然死亡的发生率为 30%,并且随着孕妇年龄的增加而增加。监测建议表明,应在妊娠 18-20 周时进行详细的超声检查和胎儿超声心动图检查,并在 28-30、34-36 和 38 周时进行进一步的超声检查,以评估是否有上消化道梗阻、乳糜胸、胎儿积水和宫内生长受限的证据。如果发现异常,则建议加强胎儿监测。如果发现异常,应通知当地儿科团队,以便他们参与计划产后护理和治疗。
先天性心脏缺陷
约 50% 的唐氏综合征患者会罹患先天性心脏病 (CHD),最常见的是 AVSD(唐氏综合征患者中 42% 的先天性心脏病)、室间隔缺损 (22%) 和房间隔缺损 (16%) 。虽然具体类型的先天性心脏病的发病率取决于年龄和种族,但主要的一点是先天性心脏病会严重影响患者的生活质量。在怀孕期间,建议进行胎儿超声心动图检查。出生后应进行心脏病学检查,并在出生后第一个月内进行另一次超声心动图检查。治疗与一般人群相同,包括手术修复。唐氏综合征儿童手术后的死亡率等于或低于一般人群。所有唐氏综合征患者都应终生每年进行一次筛查,以发现是否有获得性瓣膜疾病和心力衰竭的征兆。
睡眠呼吸暂停
阻塞性睡眠呼吸暂停在唐氏综合征患者中很常见,估计患病率为 54–90% 。每次健康检查时都应筛查症状;这些症状包括大声打鼾、呼吸沉重、夜晚不安和白天嗜睡,以及神经认知症状,如易怒、抑郁、偏执、认知下降和行为问题。建议所有 4 岁以下的唐氏综合征儿童进行整夜多导睡眠图检查,无论其症状如何。有人建议使用家庭血氧测定等替代方法识别高危人群,并减少需要全面诊断致病基因鉴定基因解码的儿童数量。睡眠呼吸暂停的治疗包括使用持续气道正压通气 (CPAP)、下颌前移装置和减肥。可以考虑手术干预,包括扁桃体切除术和腺样体切除术,尽管手术后睡眠呼吸暂停可能会持续存在。
甲状腺功能障碍
约 1% 的唐氏综合征(DS)患者患有先天性甲状腺功能减退症,据报道,50% 以上的唐氏综合征(DS)新生儿甲状腺功能检查结果异常。患甲状腺疾病的风险终生存在,并且患自身免疫相关甲状腺功能障碍的风险会随着年龄的增长而增加。由于临床诊断可能很困难,因此定期进行血液筛查非常重要。应在出生后以及 6 个月和 12 个月大时测量促甲状腺激素 (TSH) 和甲状腺素 (T4) 水平。然后应每年重复测量 TSH。
阿尔茨海默病
相当一部分唐氏综合征患者会发展为早发性 AD,这与 APP 过量生产有关,痴呆是 70% 唐氏综合征老年人的近端死亡原因。临床症状在 40 岁后出现,77% 的 60-69 岁唐氏综合征患者会出现认知能力下降,而 70 岁以上的患者中则高达 100% 会出现认知能力下降 (图 5)。需要在现有认知表型和个体风险(包括APOE基因型)的背景下理解认知能力下降,因为APOE ⋆ ε4等位基因的存在似乎会增加因 AD 导致早期认知能力下降的风险和死亡风险 (图 5b)。
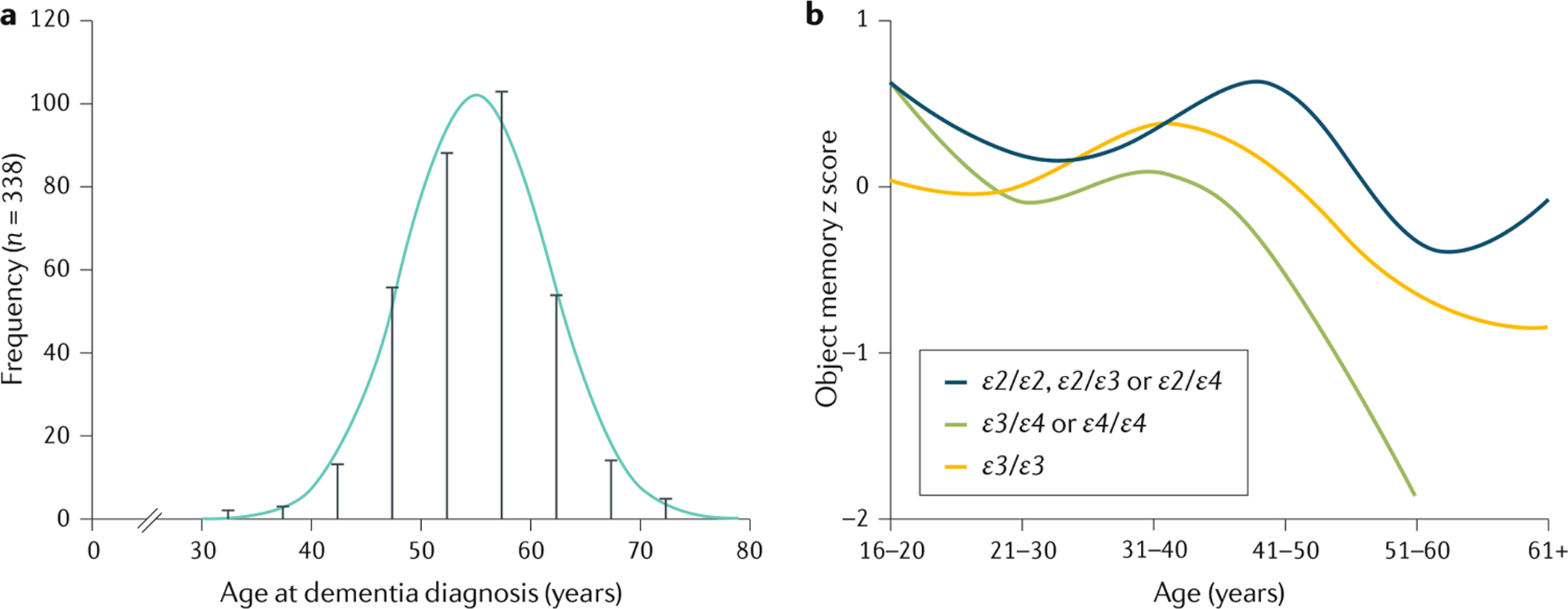
图 5:阿尔茨海默病的患病率和唐氏综合症的认知能力下降
a:唐氏综合症 (DS) 患者痴呆症诊断年龄的分布。唐氏综合症患者患阿尔茨海默病的风险与年龄密切相关。痴呆症诊断的平均年龄为 55 岁,不过有些人从 40 岁开始就出现认知能力下降,而其他人直到 60 岁以后才被诊断出来。b | 唐氏综合症患者的认知能力下降,以记忆测试的表现 ( z分数)来衡量。来自伦敦唐氏综合症联盟的横断面数据显示了唐氏综合症患者的物体记忆任务测试分数按年龄和载脂蛋白 E ( APOE ) 基因型分布。该任务是衡量短期和延迟记忆的,适合唐氏综合症患者。要求参与者通过两次立即回忆试验和一次 5 分钟后的延迟回忆试验来命名和回忆七个物体250。根据APOE基因型对数据进行拆分,以比较APOE的ε4等位基因(APOE ⋆ ε4;增加晚发型散发性阿尔茨海默病的风险)与APOE ⋆ ε2等位基因(降低 AD 风险)和APOE ⋆ ε3等位基因(对 AD 风险有中性影响)对认知表现的影响。具有APOE ⋆ ε3 / APOE ⋆ ε4或APOE ⋆ ε4 / APOE ⋆ ε4基因型的个体的认知能力从 40 岁开始下降,明显早于痴呆症诊断的平均年龄,也早于具有APOE ⋆ ε2 / APOE ⋆ ε2、APOE ⋆ ε2 / APOE ⋆ ε3、APOE ⋆ ε2 / APOE ⋆ ε4或APOE ⋆ ε3 / APOE ⋆ ε3基因型的个体。a 部分改编自 REF. 251,CC-BY-4.0(https://creativecommons.org/licenses/by/4.0/)。
唐氏综合征患者早期就会出现记忆力和注意力缺陷,就像散发性 AD 患者一样,但通常直到出现行为变化时才会被注意到。一些患有唐氏综合征和痴呆症的患者可能会出现癫痫发作,随着病情的进展,通常会出现神经系统症状,例如肌阵挛和癫痫发作。
建议所有唐氏综合征患者在 30 岁时进行认知和适应功能基线评估,以帮助将来的监测和诊断。管理应侧重于早期发现和支持措施。使用乙酰胆碱酯酶抑制剂可能会带来积极益处,尽管有些人可能会出现心率减慢和其他不良反应。唐氏综合征和痴呆症患者应获得足够的支持,因为他们的需求会随着疾病的进展而增加。应努力让患者留在熟悉的家中,并根据个人情况和需求将其转移到其他提供者。
癫痫
8% 的唐氏综合征儿童患有癫痫,发病年龄有两个高峰 (一个高峰在 3 岁之前,另一个在 30 岁之后 )。婴儿发病,最常见的是婴儿痉挛,与一种称为 West 综合征的严重癫痫有关,该综合征包括脑电图上的精神运动倒退和痉挛性心律失常 (惊厥之间的一种异常活动模式),在报告的婴儿癫痫发作中约 6-32% 有此表现。癫痫的晚期发作与 AD 的发展有关 。癫痫的治疗取决于病因,但通常涉及抗惊厥药物,这通常可有效减少癫痫发作。
听觉和视觉
传导性听力损失在唐氏综合征患者中很常见,渗出性中耳炎的患病率很高。出生后应进行基础听力测试,例如使用脑干听觉反应,然后每 6 个月进行一次,直到学龄,此后至少每年进行一次。早期识别和治疗可以降低以后听力损失的风险。感音神经性听力损失在成年期越来越常见。助听器和人工耳蜗植入治疗已取得成功。使用言语治疗、交流辅助工具和手语(包括 Makaton 语言计划)也可能有益。
由于唐氏综合征患者患屈光不正、白内障(先天性和发育性)、圆锥角膜和弱视的风险较高,因此应在出生时以及一生中定期进行眼科检查,最好每 1-2 年一次。应及时给予治疗和矫正辅助。白内障手术是常规手术,通常效果良好。
寰枢椎不稳定
约 1-2% 的唐氏综合征(DS)儿童会出现寰枢椎半脱位(第一和第二颈椎错位)。如果出现这种情况,应告知父母,参加体育运动会增加儿童脊髓损伤的风险。应使用颈椎射线照相术评估颈部疼痛、虚弱、痉挛、步态困难和反射亢进等症状。
精神健康
与普通人群相比,唐氏综合征患者的行为和心理健康问题患病率更高,尤其是抑郁症和焦虑症。一小部分患有唐氏综合征的青少年和年轻人会出现急性退化(也称为唐氏综合征瓦解性障碍),与以前的功能水平相比,他们的技能和独立性会下降。目前,这种衰退的原因尚不清楚,尽管它似乎经常在接触情绪压力源后发生。到目前为止,还没有针对这种症状的明确治疗方法。
唐氏综合征患者的精神健康问题诊断可能因智力障碍、沟通困难和非典型症状的存在而变得复杂。心理压力因素的影响不容小觑,包括住宿、学校或护理安排的转变以及丧亲之痛。精神疾病的治疗应以标准临床指南为基础,唐氏综合征患者对行为干预、精神药物治疗和心理治疗有积极的反应。
神经发育
唐氏综合征是导致智力障碍的最常见遗传原因,大多数唐氏综合征患者被归类为轻度至中度残疾。他们的认知特征表明,他们在视觉学习方面具有优势,但在表达性语言、言语工作记忆和情景记忆方面较弱。然而,认知功能存在很大差异,智商、语言、注意力、记忆力和功能能力各不相同。
唐氏综合征患者通常患有自闭症谱系障碍 (~10–15%) 和注意力缺陷多动障碍 (ADHD; ~6%),应进行适当的筛查。临床表现可能与一般人群不同,评估可能需要专家的意见。建议对 ADHD 进行标准治疗,尽管智力障碍患者对兴奋剂药物的反应可能不如没有智力障碍的人明显。
产前治疗以改善神经认知
人们越来越关注利用唐氏综合征的产前诊断作为提供产前治疗以改善神经认知的机会。唐氏综合征患者的神经发生障碍始于产前,是导致胎儿大脑生长和形状异常的几个因素之一 ,这些异常可在妊娠晚期的产前超声和 MRI 图像中识别。因此,据推测,在胎儿或新生儿早期开始的治疗效果最大
到目前为止,大多数涉及胎儿治疗的实验都是使用唐氏综合征(DS)小鼠模型进行的。产前治疗可使 Ts65Dn 小鼠的学习能力和神经行为在出生后得到改善,包括服用血管活性肠肽 NAPVSIPQ (NAP) 和 SALLRSIPA (SAL) 、母体膳食胆碱补充和抑制 DYRK1A。例如,产前使用 DYRK1A 抑制剂 EGCG 进行治疗,该药物可穿过小鼠的胎盘和血脑屏障,改善 Ts65Dn 小鼠颅骨穹窿形态缺陷的某些方面。此外,从胚胎第 10 天 (E10) 到 E15 对怀孕的 Ts1Cje 母鼠口服新型 DYRK1A 抑制剂 ALGERNON(改变神经元的生成),可以改善其幼鼠的神经发生、皮质发生和行为表现,这可能是通过增强神经干细胞的增殖来实现的。
从小鼠模型致病基因鉴定基因解码中还发现了其他潜在的治疗靶点。例如,使用二甲双胍激活 PGC1α 通路可逆转唐氏综合征人类胎儿成纤维细胞的线粒体功能障碍。唐氏综合征胎儿成纤维细胞中 PGC1α 的 mRNA 和蛋白质水平显著降低,二甲双胍治疗可增加氧消耗和细胞 ATP 浓度、改善呼吸活动、增加线粒体膜电位并逆转这些细胞中的线粒体超微结构异常。二甲双胍已获得 FDA 监管部门批准用于治疗妊娠期糖尿病,因此在怀有唐氏综合征胎儿的孕妇的临床试验中使用二甲双胍可能会面临更少的监管障碍。唐氏综合征小鼠模型致病基因鉴定基因解码还表明,产前使用选择性血清素再摄取抑制剂氟西汀治疗可恢复海马神经发生和连接、颗粒细胞数量和树突形态以及海马依赖性记忆功能。美国德克萨斯大学正在对孕妇(无精神疾病)进行高剂量氟西汀临床试验,尽管目前尚未公开有关该试验的信息。然而,在妊娠前三个月使用氟西汀可能会产生不良影响,例如增加先天性心脏病和其他畸形的风险。
芹菜素是一种天然黄酮,存在于柑橘类水果和绿叶蔬菜中,它也有望减少氧化应激,提高 21 三体综合征胎儿的人体细胞的总氧化能力,并改善产前治疗的新生儿和成年 Ts1Cje 小鼠的某些行为。认识到改善胎儿大脑发育的可能性,以及对越来越多的化合物进行测试以最终用于临床试验,使唐氏综合征(DS)致病基因鉴定基因解码成为一个非常激动人心的时刻,尽管由于证据不足而无法提出补充建议。
免疫和血液系统
唐氏综合征患者更容易感染,尤其是呼吸道感染。应及时发现和治疗感染。免疫接种没有禁忌症,应遵循标准的儿童接种时间表。
唐氏综合征患者还容易患自身免疫性疾病,尤其是乳糜泻。如果出现乳糜泻症状,建议检测组织转谷氨酰胺酶 2 的免疫球蛋白 A 抗体水平,但对无症状个体进行筛查并不划算。唐氏综合征患者中 1 型糖尿病、斑秃和 Addison 病的患病率也高于一般人群。
与没有唐氏综合征的儿童相比,唐氏综合征儿童罹患急性白血病的风险显著增加。5-30% 的唐氏综合征患者在 3 个月前会出现短暂性骨髓增生性疾病 (TMD),也称为唐氏综合征短暂性白血病,应在出生后 3 天内进行血细胞计数和胶片检查,以便识别和监测这种疾病225。虽然 TMD 通常在出生后几个月内无需治疗即可痊愈,但它似乎会增加 5 岁时患白血病的风险,据估计,20-30% 的 TMD 患者会继续发展为唐氏综合征髓系白血病。
生活质量
尽管人们认识到耻辱感和文化规范可能会成为唐氏综合征患者积极参与社区活动的障碍,但对唐氏综合征患者生活质量的致病基因鉴定基因解码仍然有限。如果在生活中得到充分的支持,许多唐氏综合征患者可以相当独立地生活。唐氏综合征患者家庭成员的观点已经得到一定程度的致病基因鉴定基因解码。支持团体和唐氏综合征网络可以提供宝贵的意见,唐氏综合征患者及其家人应该了解当地和国家组织。
如今,唐氏儿童在主流学校接受常规教育,通常还会获得一对一或针对特殊教育需求的额外支持。一些唐氏儿童在融合教室接受教育,而不是在完全独立的教室接受教育,可以在语言和读写能力方面获得额外的好处,而其他一些儿童可能需要专科学校。
许多唐氏综合征患者都有工作,并且报告了相当高的满意度,许多人已经成为成功的专业人士,包括艺术家、演说家和演员。然而,希望工作的唐氏综合征患者的数量与已经找到工作的唐氏综合征患者的数量之间存在差距。他们可能会从事志愿工作,这似乎比一般成年人口更常见。文化假设、社会护理削减和工作调整不足可能会使唐氏综合征患者难以就业。
生活质量会受到身体健康的影响,而积极的筛查计划和早期干预可以减少长期住院和疾病带来的负面影响。识别和纠正听力和视力问题可以减少对交流和发展的负面影响。肥胖、颈椎异常和肌肉问题可能会导致唐氏综合征患者无法进行体育锻炼,在美国,唐氏综合征儿童的体育锻炼水平低于同龄人234。然而,如果有机会,大多数人都可以参加锻炼计划,这对健康和幸福有积极的影响。
由于早期认知能力下降,许多患有唐氏综合症的成年人随着年龄增长需要的支持也随之增加。充足的主动支持、定期重新评估需求以及为预测功能障碍而设立的服务,对于他们的生活质量至关重要。
外表
尽管随着医疗技术的进步和社会变革的益处,唐氏综合征患者的生活得到了很大改善,但目前尚无基于对唐氏综合征分子病理生理学的现有理解的唐氏综合征治疗方法。因此,为了设计有效的治疗方法,需要进一步致病基因鉴定基因解码以加深对该综合征每种症状和特征的生物学理解。下面讨论了进一步致病基因鉴定基因解码的最重要主题。
了解遗传变异对唐氏综合征不同且多变的表型特征和表现的贡献是一个重要目标,这一目标可以通过对数千名唐氏综合征患者进行基因组测序,并将遗传变异与每个个体的详细表型特征联系起来来实现。这项工作基于一个合理的假设,即基因组差异是广泛表型变异的主要决定因素。此外,需要识别出以三个拷贝促成唐氏综合征表型的基因和其他功能性基因组元素。有支持证据表明,导致单倍体不足疾病状态的基因(其中一个功能性拷贝是不够的)在以三个拷贝存在时也可能很重要,唐氏综合征就是这种情况。例如,DYRK1A是单倍体不足的(功能丧失不耐受概率 (pLI) 评分为 1),丢失一个DYRK1A拷贝会导致智力障碍。多项致病基因鉴定基因解码还表明, DYRK1A的额外拷贝与 21 三体综合征导致的生物功能障碍有关。但 pLI 评分可能不是与唐氏综合征(DS)病理生理学有关的基因的绝对鉴别因素。例如,APP与 DS患者的 AD 密切相关,但其 pLI 评分为 0.06,与单倍体充足基因相符。此外,与21 三体综合征转录失调有关的基因HMGN1 ,其 pLI 评分为 0.22。除了特定基因对 HSA21 的致病作用外,还需要致病基因鉴定基因解码染色体外物质的表型贡献,无论其基因含量如何。
需要使用实验模型(例如小鼠和其他生物7 )来致病基因鉴定基因解码处于不同发育阶段的大脑和其他器官。单细胞技术的进步使得能够致病基因鉴定基因解码单个细胞中的基因组、转录组、组蛋白修饰、染色质接触和蛋白质水平,这必将为唐氏综合征(DS)中改变的发育途径的细胞机制以及染色质构象对基因表达失调的影响提供见解。
为改变 HSA21 和其他染色体上基因表达失调而进行的实验努力可能会产生值得进一步探索的新治疗方法。例如,通过在 HSA21 的一个拷贝上引入 X 染色体失活效应物XIST来实验性地沉默大片染色体区域,或沉默HMGN1、DYRK1A或APP三个等位基因之一,可能会挽救唐氏综合征的一些表型特征。
唐氏综合征患者的早发性 AD 在遗传学上与某些形式的常染色体显性 AD 类似,由于APP三倍化,淀粉样蛋白通路与 AD 病理密切相关。因此,应在唐氏综合征患者中试用针对淀粉样蛋白的药物,因为他们患早发性 AD 的风险非常高,最好在出现任何症状之前进行,以预防或延缓痴呆症的发作。脑内淀粉样蛋白或 tau 沉积物的 PET 成像或脑脊液中 Aβ 肽比率的测量已被证明是可行的患者分层生物标志物,而神经退行性疾病标志物神经丝轻链和神经元五聚蛋白 2 的血浆水平有望成为治疗反应的生物标志物。
针对唐氏综合征患者开展基于人群的纵向大规模队列致病基因鉴定基因解码将有助于进一步了解唐氏综合征患者目前存在的并发疾病、家庭态度、教育成就、个人经历和社会成就。日益复杂的分析方法与改进的遗传模型相结合,为更多有前景的疗法的转化带来了希望。
(责任编辑:佳学基因)
