












【佳学基因基因检测】人类遗传病基因检测所需要的序列、结构与功能对致病基因的判断与评估
遗传病致病基因鉴定基因解码基因检测编制了一个由计算机程序预测或从 UniProt 特征字段中检索的蛋白质序列、结构和功能特性数据集。对于任何一个功能特性改变来说,与所有人类蛋白质中该特性的背景频率相比,采用富集对数比分来确定具有致病性单氨基酸变异SAV的氨基酸位置是否有任何特性被富集或被剔除。遗传病的致病基因鉴定基因解码基因检测观察到致病性致病性单氨基酸变异SAV 中保守位置富集了 1.7 倍,可变位置减少了 3 倍以上。类似地,三个紊乱预测程序(DISOPRED3、SPOT-Disorder 和 IUPred2A)的结果一致表明,致病性单氨基酸变异SAV在有序区域富集,无序区域剔除。预测的 β 链和 α 螺旋在致病性致病性单氨基酸变异SAV中略微偏爱,而二级结构预测的卷曲区域略微倾向于剔除。预测的二级结构类型的富集/耗尽并不大(小于 1.33 倍)。先前基于已知结构对二级结构偏差的研究也表明,致病变异和良性变异的二级结构分布之间几乎没有差异,尽管作者发现了螺旋/转弯/桥的微弱富集和 β- 的微弱耗尽致病变异链。不同的发现可能是由于数据集(本研究中的全长蛋白质与具有已知结构的区域)和二级结构估计方法(预测与实际)的差异。遗传病的基因序列突变与结构功能变化的关系研究还观察到致病性单氨基酸变异SAV中低复杂性区域和卷曲螺旋区域的轻微耗尽。
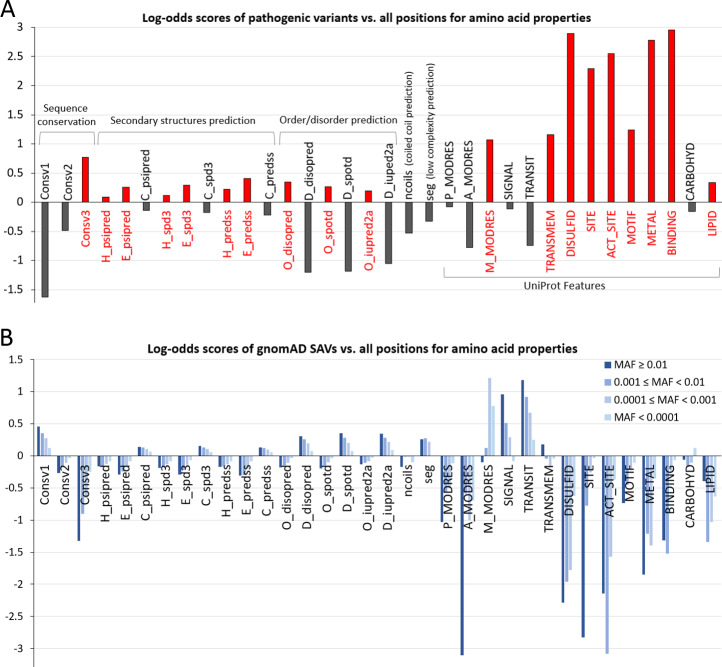
A) 与所有氨基酸位置相比致病性 SAV 特性的富集/耗尽(y 轴显示基于 log2 的对数比值分数)。符号:Consv1——保守分数低的位置(0 到 0.3 之间),Consv2——保守分数中等的位置(0.3 到 0.6 之间),Consv3——保守分数高的位置(大于 0.6)。 H_psipred、E_psipred、C_psipred 是 PSIPRED 对 α-螺旋、β-链和螺旋的二级结构预测。二级结构预测程序 SPIDER(H_spd3、E_spd3、C_spd3)和 PREDSS(H_predss、E_predss、C_predss)使用相同的符号。 O_disopred 和 D_disopred 分别对应 DISOPRED 预测的有序和无序区域,无序预测程序 SPOT-DISORDER(O_spotd 和 D_spotd)和 IUPRED2A(O_iupred2a 和 D_iupred2a)使用相同的符号。符号 ncoil 和 seg 分别是预测的盘绕线圈区域和低复杂度区域。 P_MODRES、A_MODRES 和 M_MODRES 分别是在 UniProt 中被注释为被磷酸化、乙酰化和甲基化修饰的位置。 SIGNAL、TRANSIT 和 TRANSMEM 是 UniProt 中注释为信号肽、转运肽和跨膜片段的位置。 DISULFID、SITE、ACT_SITE、MOTIF、METAL、BINDING、CARBOHYD 和 LIPID 是 UniProt 特征字段中这些关键词中注释的位置。 B) 与所有氨基酸位置相比,具有不同 MAF 范围(从浅蓝色到深蓝色:MAF < 0.0001、0.0001 ≤ MAF < 0.001、0.001 ≤ MAF < 0.01、0.01 ≤ MAF)的 gnomAD SAV 中氨基酸特性的富集/消耗.
对于具有亚细胞定位迹象的区域,信号肽和线粒体转运肽在致病性 SAV 中被耗尽,但跨膜片段丰富了 2 倍以上。源自 UniProt 特征的几个特性在致病性 SAV 中表现出贼强的富集。它们与蛋白质稳定性(UniProt 特征 DISULFID:参与二硫键的半胱氨酸残基)或功能(UniProt 特征:SITE、ACT_SITE、METAL、MOTIF 和 BINDING)有关。除 MOTIF 特征外,它们在致病性 SAVs 中表现出超过 4 倍的富集(基于 log2 的优势得分超过 2)。
遗传病与罕见病基因解码基因检测还分析了 gnomAD(基因组聚合数据库)数据库中超过 12,000 个外显子组(>24,000 个等位基因)中发现的 SAV,该数据库提供了来自一般人群的自然变异的综合分类。常见的单氨基酸变异SAV(MAF ≥ 0.01)应该大部分是良性的,它们只占 gnomAD单氨基酸变异SAV的一小部分(4,885,239 中的 27,813,约 0.57%)。 gnomAD 数据库拥有更多罕见的单氨基酸变异SAV,MAF 小于 0.01,其中很大一部分是单体(在所有外显子组中只发现一次)。单氨基酸变异SAV根据它们的 MAF 将 gnomAD单氨基酸变异SAV分为四类(MAF < 0.0001、0.0001 ≤ MAF < 0.001、0.001 ≤ MAF < 0.01 和 MAF ≥ 0.01)。大多数单氨基酸变异SAV(4,885,239 中的 4,588,805,约 94%)属于 MAF < 0.0001 的稀有单氨基酸变异SAV类别,而约 4.4%(27,813)和 1.1%(53,489)属于 0.0001 ≤ MAF <0.001 和 0.001 类别分别≤ MAF < 0.01。人口瓶颈事件可能是常见单氨基酸变异SAV耗尽的部分原因,而贼近历史上人口的爆炸性增长可能导致稀有单氨基酸变异SAV数量过多。
以与致病性单氨基酸变异SAV相同的方式分析每个 gnomAD单氨基酸变异SAV类别中蛋白质序列、结构和功能特性的富集。与致病性单氨基酸变异SAV相比,常见的 gnomAD单氨基酸变异SAV(MAF ≥ 0.01)通常表现出相反的富集/消耗趋势。 DISULFID、SITE、ACT_SITE、METAL、MOTIF 和 BINDING 等属性在常见的 gnomAD单氨基酸变异SAV中表现出贼显着的消耗,在致病性单氨基酸变异SAV中表现出贼强的富集。相比之下,常见单氨基酸变异SAV中丰富的特性包括可变位置 (Consv1)、二级结构预测的线圈区域、预测的无序区域、低复杂性区域、信号肽和线粒体转运肽。当从常见的 gnomAD单氨基酸变异SAV类别移动到不太频繁的 gnomAD单氨基酸变异SAV类别时,属性的丰富或减少逐渐减少。这种行为表明,许多低频单氨基酸变异SAV,尤其是一般人群中 MAF 小于 0.0001 的那些可能是有害的,因为功能上重要的残基(由 UniProt Features SITE、ACT_SITE、BINDING、METAL 和 MOTIF 指定的属性)在这些罕见的单氨基酸变异SAV比普通的单氨基酸变异SAV更重要。
(责任编辑:佳学基因)